- Software
- Open access
- Published:
Bio::Homology::InterologWalk - A Perl module to build putative protein-protein interaction networks through interolog mapping
BMC Bioinformatics volume 12, Article number: 289 (2011)
Abstract
Background
Protein-protein interaction (PPI) data are widely used to generate network models that aim to describe the relationships between proteins in biological systems. The fidelity and completeness of such networks is primarily limited by the paucity of protein interaction information and by the restriction of most of these data to just a few widely studied experimental organisms. In order to extend the utility of existing PPIs, computational methods can be used that exploit functional conservation between orthologous proteins across taxa to predict putative PPIs or 'interologs'. To date most interolog prediction efforts have been restricted to specific biological domains with fixed underlying data sources and there are no software tools available that provide a generalised framework for 'on-the-fly' interolog prediction.
Results
We introduce Bio::Homology::InterologWalk, a Perl module to retrieve, prioritise and visualise putative protein-protein interactions through an orthology-walk method. The module uses orthology and experimental interaction data to generate putative PPIs and optionally collates meta-data into an Interaction Prioritisation Index that can be used to help prioritise interologs for further analysis. We show the application of our interolog prediction method to the genomic interactome of the fruit fly, Drosophila melanogaster. We analyse the resulting interaction networks and show that the method proposes new interactome members and interactions that are candidates for future experimental investigation.
Conclusions
Our interolog prediction tool employs the Ensembl Perl API and PSICQUIC enabled protein interaction data sources to generate up to date interologs 'on-the-fly'. This represents a significant advance on previous methods for interolog prediction as it allows the use of the latest orthology and protein interaction data for all of the genomes in Ensembl. The module outputs simple text files, making it easy to customise the results by post-processing, allowing the putative PPI datasets to be easily integrated into existing analysis workflows. The Bio::Homology::InterologWalk module, sample scripts and full documentation are freely available from the Comprehensive Perl Archive Network (CPAN) under the GNU Public license.
Background
In recent years, large protein-protein interaction (PPI) datasets have allowed the description of relationships between proteins in complex biological systems [1]. These data are commonly derived from yeast two hybrid (Y2H), co-immunoprecipitation or tandem affinity purification (TAP) assays and have been obtained from a variety of unicellular and multicellular organisms [2–6]. Recent advances in high resolution mass spectrometry have further contributed to the rapid accumulation of PPI data [7–9]. Unfortunately, large scale experimental discovery of PPIs remains difficult, expensive and beyond the means of many experimentalists. Currently, PPI data is almost exclusively limited to a few popular model organisms and amongst these coverage of the captured interactions is often biased to a particular domain and incomplete. For many organisms, PPI data lags behind or is non-existent compared with genome sequence data.
In an attempt to address the relative paucity of data, a number of computational techniques have been proposed to predict and prioritise PPIs [10, 11]. While the number of such methods is large, we focus here on methods that transfer functional information using cross-species orthology projection [12, 13]. In essence, the rationale is that for interacting proteins x and y in organism  we expect (under certain conditions) that their orthologues x' and y' in organism
we expect (under certain conditions) that their orthologues x' and y' in organism  will also interact. Such conserved interaction pairs are called 'interologs' [14, 15]. The potential use of interolog mapping has been explored in several organisms including Homo sapiens[16–22], Helicobacter pylori[23], Saccharomyces cerevisiae[24], Plasmodium falciparum[25] and Magnaporthe grisea[26]. Additionally a number of quantification methods have been developed to assess the confidence of predicted interologs [27–29].
will also interact. Such conserved interaction pairs are called 'interologs' [14, 15]. The potential use of interolog mapping has been explored in several organisms including Homo sapiens[16–22], Helicobacter pylori[23], Saccharomyces cerevisiae[24], Plasmodium falciparum[25] and Magnaporthe grisea[26]. Additionally a number of quantification methods have been developed to assess the confidence of predicted interologs [27–29].
Several web interfaces to interolog databases have been developed (e.g. HomoMINT [19] and Ulysses [20]), but these are essentially ad hoc efforts. They consider a small set of organisms for interolog prediction in restricted biological domains, thus hindering more widespread use. Underlying data sets are often frozen at the moment of publication or curated for a limited period of time (e.g. InteroPORC [28]), are dependent on other projects that are not updated (e.g. Integr8 [30]) or are based on algorithms that are not state-of-the-art. Considering that both orthology projection methods and interaction data are continually updated, such static databases are destined to obsolescence. To date the only project that provides the option to discover interologs with up to date data is OpenPPI predictor [31]. This represents a step forward, but relies on the user providing both the orthology relationships and known PPI data and only performs mappings between two species, without ranking or prioritising the putative PPI network obtained.
To address the lack of tools for performing multi-species interolog prediction 'on the fly' we created the Perl module Bio::Homology::InterologWalk. The tool relies on BioPerl [32], the Ensembl Perl Core and Compara APIs [33, 34] and the EBI-Intact PSI Common Query InterfaCe (PSICQUIC) enabled web service [35, 36] for its operation. Bio::Homology::InterologWalk is freely available under the GNU General Public Licence at the Comprehensive Perl Archive Network (CPAN) [37, 38].
Bio::Homology::InterologWalk accepts as input a list of Ensembl gene accession numbers from any of the vertebrate or metazoan genomes in Ensembl and also for all species in the Ensembl pan-taxonomic Compara database. The tool searches the Ensembl Core and Compara databases and the PSICQUIC-enabled EBI-Intact PPI database to collect and analyse gene orthology and PPI data, together with ancillary information. It then provides the option of filtering the putative interactions to retain those with strong experimental or phylogenetic support. In addition the user can query the PPI database directly to collect all known interactions for the input gene list. This allows the ready comparison of putative PPIs from interolog projection to known PPIs. The software outputs plain text tab-separated files and can also output network representations of the PPI data and their attributes in a format compatible with the widely used biological network analysis tool Cytoscape [39].
We demonstrate the use of the software to investigate the potential of interolog projection on the genome of the fruit fly, Drosophila melanogaster[40]. The analysis (a) generates a novel putative PPI network that strengthens the connectivity of the known PPI network (b) proposes new interaction candidates. We further calculate an Interaction Prioritisation indeX (IPX) for each of the PPIs and use these to create a sub-network centred on a core of 10 DNA replication proteins.
Implementation
Overview
A high-level schematic describing our implementation of the interolog walk concept is shown in Figure 1. The main purpose of Bio::Homology::InterologWalk is to obtain a list of putative PPIs given a set of user-selected gene identifiers in one genome of interest. In order to be compatible with the module, the initial dataset must be a list of Ensembl IDs belonging to species in Ensembl Vertebrates, EnsemblGenomes Metazoa or Ensembl Pan-taxonomic Compara databases.

The Bio::Homology::InterologWalk concept. Schematics illustrating the principle behind interolog mapping. Proteins x and y are known to interact in a reference genome. If they have orthologues x' and y' in the genome of interest, under certain conditions the existence of a putative x' - y' interaction can be assumed. Bio::Homology::InterologWalk implements this in a three-steps algorithm. 1. get orthologues of genes of interest in reference genome(s). queries the initial gene list against one or more Ensembl databases and retrieves their orthologues. Options can be set to specify stringency of retrieved hits. Ancillary data fields are computed. 2. get interactions in reference genome(s). queries the orthology list built in (1.) against PSICQUIC-enabled PPI databases using REST. This step will enrich the dataset built in (1.) with the interactors of those orthologues, if any, plus ancillary data -- including parameters describing the nature and origin of the annotated interaction. 3. get orthologues from reference genome(s) back in genome of interest. In this step the interactor list built in (2.) is queried against one or more Ensembl databases (again using the Ensembl Perl API) to find orthologues back in the original genome of interest. As in (1.), a number of supplementary information fields are computed.
To carry out an interolog walk, Bio::Homology::InterologWalk will first query the gene identifiers chosen by the user against the Ensembl databases using the Ensembl Compara API [41], retrieving a list of orthologous gene IDs. Next, the algorithm will use the Representational State Transfer (RESTful) interface [42] to interrogate a PSICQUIC-compliant PPI database with the list of orthologues returned by Ensembl, to retrieve the list of known PPIs involving them. While there are already several interaction databases implementing the PSICQUIC interface [43–50], Bio::Homology::InterologWalk currently relies on EBI IntAct [36] as its source of experimental interactions. Having obtained a list of interactors for the orthologues of the initial gene set, in the last step of the main data mining procedure Bio::Homology::InterologWalk will project the interactions retrieved (again, using the Ensembl Compara API) back to the original species of interest. The final output is a list of putative interactors for the initial gene set and several fields of supporting data for the forward orthology map, the PPI data collection, and the backward orthology map. These metadata fields can be analysed by a sub-module of the tool, to calculate a prioritisation index for the predicted PPIs (Figure 2A).

Supplementary Data Fields & Prioritisation Features. Schematics summarizing the features used to prioritise the resulting putative PPIs. For each PPI, a number of metadata fields are collected during the three main steps of the algorithm. Two metrics can optionally be computed: an Interolog Prioritisation Index (IPX) and a PPI Conservation Score (PCS) (Additional File 2 - Definitions). A. Overall view showing the contributing data fields. B-E. Sample prioritisation features. B: Phylogenetic distances (according to TreeBeST). For each of the two orthologous pairs, a node-to-node distance (nnD
i
) and two distances from the First Shared Ancestor  are computed. C: experimental interaction observed in multiple taxa - a component of the IPX is proportional to the number of reference genomes contributing to a putative PPI evidence. D: experimental interaction reconfirmed through multiple detection methods - a component of the IPX is proportional to the number of detection methods used to obtain experimental PPI evidence in the reference genome. E: PPI Conservation Score. The conservation score depends on (1) the density of the most-connected γ -clique that includes x, y and their mutual interactors and (2) the number of edges of the γ clique.
are computed. C: experimental interaction observed in multiple taxa - a component of the IPX is proportional to the number of reference genomes contributing to a putative PPI evidence. D: experimental interaction reconfirmed through multiple detection methods - a component of the IPX is proportional to the number of detection methods used to obtain experimental PPI evidence in the reference genome. E: PPI Conservation Score. The conservation score depends on (1) the density of the most-connected γ -clique that includes x, y and their mutual interactors and (2) the number of edges of the γ clique.
The procedure is organised as a pipeline of related data-processing activities. The output of the basic pipeline can be further processed with the help of other methods in the module: it is possible to scan the results and compute counts, check for duplicate entries, isolate new gene IDs (not part of the original dataset) and generate Cytoscape-compatible network representations of the data. The package documentation gives a detailed account of post-processing options.
An additional stand-alone functionality of the module is the direct PPI pipeline: it is possible to use Bio::Homology::InterologWalk to mine all the experimental PPIs involving the initial gene list in the genome of interest (without mapping to reference genomes using orthology). This dataset is a 'snapshot' of the current experimental PPI network for the input dataset. As such it is useful both by itself, because it tells what is currently know about the PPIs involving the initial genes, and as a term of comparison for the putative PPI - because it can be used to evaluate the amount of overlap between the known and putative networks, as well as the novelty of the putative data. Additional file 1: 'Simplified schematics of the Interolog Walk Pipeline' illustrates both the putative and the experimental pipeline in detail.
One of the defining features of Bio::Homology::InterologWalk is that the retrieval of both orthology data and protein interaction data happens on-the-fly. The user inputs a list of gene IDs plus a number of set-up parameters, and the data will be downloaded through web-service interfaces each time the program is run.
Data Sources
Orthology Predictions from Ensembl Compara
Bio::Homology::InterologWalk uses the Ensembl Perl API http://www.ensembl.org/info/data/api.html to access the comparative biology data provided by the Ensembl Project through Ensembl Compara. The orthology prediction method used by Ensembl Compara is described by Vilella et al. [41] and identifies several classes of homology association between genes (Additional file 2: 'Definitions').
PPI Data from EBI-IntAct
Bio::Homology::InterologWalk currently uses EBI IntAct [36] as its source of experimental interactions. As of June 2011, v. 1.1.7 of the IntAct database contains more than 267,000 curated binary protein interaction evidences [36]. Bio::Homology::InterologWalk queries Intact using the RESTful-based PSICQUIC [Aranda et al., in preparation] implementation and retrieves data in PSI-MI MITAB25 tab-delimited format [35] (Additional file 2: 'Definitions').
Options for Prioritisation of Putative Interactions
Filtering
Depending on the size of the input dataset and on the amount of information available through homology mapping, Bio::Homology::InterologWalk can produce large numbers of putative interactions. In such cases it might be beneficial to filter and prioritise these to generate a smaller set of putative interactions for further study. The Bio::Homology::InterologWalk module is composed of a number of functions that can be executed in sequence to create pipelines for retrieving interologs. Each of these functions offers options to filter the types of orthologues and interactions that are retrieved.
-
1.
Spoke interactions: the user can choose whether to return any 'spoke' interactions when using interaction retrieval functions. Spoke interactions are binary interactions that are inferred from a complex of proteins that have been isolated together and as such the evidence for the interaction is indirect. Several of the most widely used protein-protein interaction data repositories including the two largest IntAct and BioGrid explicitly draw the user's attention to the presence of spoke (or co-existence) interactions and provide the option of excluding them at an early stage.
-
2.
One-to-one orthology: for each of the orthology mapping functions the user can choose whether to restrict the mapping to explicit 1:1 relationships. This is likely to significantly reduce the number of orthologues retrieved as the evolutionary distance between mapped species increases. Restricting mappings to direct orthologues increases the likelihood that the mapped proteins retain some common functionality. Conversely considering 1-to-many or many-to-many relationships that have arisen through duplication events risks connecting proteins and interactions whose functions have diverged [51, 52].
-
3.
Experimental interactions: the user can specify whether to restrict the interactions retrieved to those that have been identified by experimental methods rather than by inference or prediction (Additional File 2: 'Definitions').
-
4.
Physical interactions: the user can choose to retrieve only those interactions that test for direct physical association between proteins (Additional File 2: 'Definitions').
Prioritisation
We have created an Interaction Prioritisation indeX (IPX) and a PPI Conservation Score (PCS) that can optionally be used to aid in the prioritisation of putative interactions.
The IPX summarises the contribution of several pieces of heterogeneous information that are collected during orthology projection and interaction retrieval. It is not intended to be a quantitative measure of interaction reliability, but rather an integration of biological information such as orthology type, phylogenetic distance (FSA), percentage identity (OPI), interaction type and detection method (including multi-method). This is similar to the work of Huang et al. [22] and Yu et al. [27]. Yu et al. used sequence similarity between the orthologous proteins to build a join similarity score, while Huang et al. proposed a scoring framework based on GO functional annotation, domain information, tissue specificity and sub-cellular localisation to rank interolog-based human putative PPIs obtained from six eukaryotes. Some of the indicators evaluated to create the IPX are:
-
Orthology Type. The kind of orthology relationship existing between an ID in the genome of interest and its orthologue in the reference genome. This feature indicates if there is a one-to-one mapping of orthologues, or if in-paralogy events in one or both sides mean we are considering a one-to-many, many-to-one or many-to-many orthologous mapping. As explained in the filtering section, we particularly value putative PPIs where both orthology relationships are of the one-to-one kind. It has been shown [51] that gene duplication is correlated with sub-functionalisation and neo-functionalisation. When the two orthologous pairs in the interolog walk are of the one-to-one kind we set a boolean variable, Θ, to a non-negative value in the score. We set Θ = 0 otherwise.
-
Expanded Complex. Indicates whether the binary interaction has been extracted from a complex using the spoke expansion model. A boolean non-negative term, ∑, is added to the score to reward each true binary interaction. ∑ = 0 for spoke-expanded binary interactions.
-
OPI. Overall Percentage Identity. A numerical index representing the percentage identity of the conserved columns between the two orthology members' sequences. Given N total samples, we define a Joint OPI as the geometric mean of the two OPIs (forward and backward orthology projection)
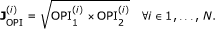
-
Node to Node Distance. A numerical indicator of the node-to-node distance in the consensus phylogenetic/species tree built by Ensembl Compara using Genetrees [41] (Figure 2B). We consider
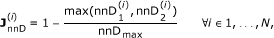
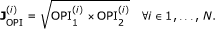
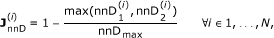
where nnD1 is the node-to-node distance between the two orthologues in the forward projection, nnD2 is the node-to-node distance between the two orthologues in the backward orthology projection and we set

-
Interaction Type & Interaction Detection Method. Features based on PSI-MI controlled vocabulary terms indicating, respectively, the type of interaction and the detection method used, within the HUPO PSI-MI hierarchy (Additional File 2: 'Definitions', Table S1).
-
PPI obtained with Multiple Methods & annotated in Multiple Organisms. Experimental PPIs reconfirmed through the usage of further detection methods and/or observed in multiple reference genomes are acknowledged by this feature (Figures 2C and 2D).
Overall, the putative PPI Interolog Prioritisation indeX is

In this expression,


agglomerates the terms relative to the PPI in the reference organism: i is a feature scoring the interaction type and d is a feature scoring the interaction detection method. mdm acknowledges those experimental PPIs present in the database more than once, with different detection methods (Figure 2D). mtaxa is set to the number of reference genomes that possess an experimental interaction projecting back to the same putative PPI (Figure 2C). The four features are normalised to make sure their values are comparable. Normalisation constants are explained in Additional File 2: 'Definitions'. The terms relative to the two orthology projections are combined in SORT:

We set ω i ,= ω o = 1. Optimisation of these two weights based on training data will allow to reward either the interaction component or the orthology component of the score to optimise performance on a case-by-case basis. Lastly, ∑ and Θ are boolean terms and we set ∑ = 0 whenever the putative PPI has been inferred from a binary PPI derived from a spoke-expanded complex (∑ = n, where n > 0 is an integer, otherwise), while Θ = n whenever the putative PPI has been inferred based exclusively on one-to-one orthology paths (Θ = 0 otherwise).
∑ and Θ are boolean flags not normalised in the IPX expression. This is done to obtain a gross selection of putative PPI samples based on co-orthology/no co-orthology and spoke/no spoke information, prior to looking at other secondary metadata features. The value n was chosen to be the smallest integer bigger than the maximum spread of the distribution of the normalised IPX features. The IPX is composed of 6 features, f = [i, d, m dm , mtaxa, JOPI, JnnD], where 0 ≤ fi ≤ 1, ∀ i ∈ 1, ..., 6 and so n = 7.
Allowing Θ and ∑ to be one order of magnitude bigger than other IPX features means the IPX distribution will take a roughly three-modal shape, depending on the combinatorial values of ∑ and Θ, as follows:
-
1.
∑ = 0, Θ = 0 (Low Tier) - the experimental interaction is spoke-expanded and at least one of the two orthology projections is not one-to-one.
-
2.
(∑ = n, Θ = 0) Θ (∑ = 0; Θ = n) (Mid Tier) - either the experimental interaction is spoke-expanded or at least one of the two orthology projections is not one-to-one.
-
3.
∑ = n, Θ = n (High Tier) - the experimental interaction is not expanded from a spoke-complex and the orthology projections are both one-to-one.
Visual inspection of the modes in the IPX distribution can be used as strategy to filter out different sets of putative PPIs, depending on the dataset considered and on the distribution of samples within the modes of the histogram. The choice of n provides good visual separation of the modes in the IPX distribution to facilitate inspection. We refer to the module code for further details.
The PPI Conservation Score (PCS) focuses on the potential for evolutionary conservation in the projected PPI by examining the density of the sub-network from which each experimental PPI is extracted. It has been shown that the connectivity of well-conserved proteins in PPI networks is negatively correlated with their rate of evolution [53, 54] and, as a consequence, more connected proteins evolve at lower rate because they are subject to higher pressure to co-evolve with interacting proteins. The PPI conservation score quantifies the degree of connectivity of the sub-network to which each experimental (known) PPI used for the interolog walk participates (Figure 2E). A binary interaction part of a very well-connected sub-network in the reference genome is more likely to have retained its functional characterisation after the projection to the organism of interest. In our implementation, we follow the method suggested by Huang and colleagues [22], and define the PPI Conservation Score as

where γ = 2 · E/[N · (N - 1)] and N and E are, respectively, the number of nodes and edges in the sub-network. Since the γ-connectedness measure is biased towards maximally connected small sub-networks, is relaxed by weighting it with the number of edges E.
Schematics illustrating the IPX and PCS are shown in Figure 2A-D and 2E respectively, and a detailed description of both can be found in Additional File 2: 'Definitions'.
There are many ways that an interolog could be prioritised. We aim for Bio::Homology::InterologWalk to be compatible with a diverse range of data and useful for many different kinds of users. Any prioritisation metric will be context-dependent and for this reason we offer a number of options to configure the process to suit the users requirements and the coverage and quality of the data available to them. As such the generalised and customisable prioritisation scheme we provide here should provide the necessary exibility to allow application across a broad range of biological domains.
Results and Discussion
Validation
Retrieving known interactions through orthology walking
We tested the functionality of the Bio::Homology::InterologWalk package by recovering known interactions using the orthologue walking principle (Figure 3). To identify known interologs for the validation analyses, we obtained the complete genomes for five well-annotated species (human, mouse, yeast, fly and worm) from Ensembl V. 61. Then, we extracted all the known experimental protein-protein associations for each of the five genomes G
i
(i = 1, ..., 5) from EBI-Intact. We define  to be the set of the
to be the set of the  experimental protein-protein interaction pairs in G
i
:
experimental protein-protein interaction pairs in G
i
:

Validation Procedure. Schematics showing the rationale for the creation of the known positive sets  for validation. (A) Complete PPI datasets for two genomes G
i
and G
j
are retrieved. Only PPIs conserved across the two species through orthology are retained. PPI pairs in G
i
satisfying this property constitute the known positive set
for validation. (A) Complete PPI datasets for two genomes G
i
and G
j
are retrieved. Only PPIs conserved across the two species through orthology are retained. PPI pairs in G
i
satisfying this property constitute the known positive set  . (B) PPI information between the IDs in is assumed unknown. (C) The gene IDs in are the input for Bio::Homology::InterologWalk. (D) The putative PPI set obtained is compared with the experimental interaction known positive set.
. (B) PPI information between the IDs in is assumed unknown. (C) The gene IDs in are the input for Bio::Homology::InterologWalk. (D) The putative PPI set obtained is compared with the experimental interaction known positive set.

Next, we selected five pairwise genome combinations G
i
G
j
: mouse-human, human-yeast, human-fly, fly-yeast and yeast-worm. For each G
i
G
j
, we define the Known Positive Evidence dataset  as the following subset of
as the following subset of  :
:

where ortho(·) is the orthology operator. is the set of all binary PPIs in G
i
that match through orthology in G
j
(Figure 3A).
The gene IDs in the five PPI sets in  were used as input for the module. To validate the ability of Bio::Homology::InterologWalk to recover known interologs (Figure 3B-D), we compared the degree of overlap between predicted nodes (gene IDs) and edges (PPIs) and known positive nodes and edges, for each of the five sets (Figure 4). For each Venn diagram, the grey set represents the known positive set , while the white set corresponds to the algorithm's predictions.
were used as input for the module. To validate the ability of Bio::Homology::InterologWalk to recover known interologs (Figure 3B-D), we compared the degree of overlap between predicted nodes (gene IDs) and edges (PPIs) and known positive nodes and edges, for each of the five sets (Figure 4). For each Venn diagram, the grey set represents the known positive set , while the white set corresponds to the algorithm's predictions.

Known Positive Set and Algorithm Prediction Overlap. Venn diagrams showing, for five representative species-pair combinations, the overlap between known positive sets (grey circle) and Bio::Homology::InterologWalk predicted set (white circle). In all observed cases, the algorithm completely rescues the known positive samples and, in addition, proposes new potential interactions and interaction candidates. The new predictions account for a minimum of 53% to a maximum of 90% of the total IDs produced and a minimum of 73% to a maximum of 96% of the total PPIs. The results suggest that even in the case of well studied organisms - provided that the hypothesis of functional conservation between orthologues is correct - most physical protein associations are still unknown.
Bio::Homology::InterologWalk successfully retrieves 100% of the positive PPIs in all cases considered. In addition, putative PPI predictions are retrieved, which are candidates that can be prioritised for experimental evaluation. Interestingly, the known positive sets appear smaller than might be expected between closely related organisms like human and mouse. This might be due to a combination of factors such as (1) the parameters for orthology classification used by Ensembl are very stringent, (2) there are biases in experimental research across organisms (the bulk of experimental predictions in each of the two species might come from experiments in different cellular domain and sub-systems) (3) experimental PPI data will likely contain false positive interactions, which will not map through orthology.
It is also interesting to note that in the case of the yeast-worm pair (Figure 4I and 4J) the number of novel IDs and novel PPIs retrieved is one order of magnitude smaller than in the other four cases. This is consistent with the relatively limited amount of experimental PPI data available for C. elegans.
Assessing the IPX using Receiver Operating Curves (ROC)
Using the known positive datasets in KP from the previous section we next calculated ROC curves to assess the performance of the IPX for each of the five species pairs. For all five datasets, the area under the curve AUC > 0.5 (Figure 5), demonstrating that there is a positive relationship between known positives and the IPX. It is important to note, however, that this correlation varies depending on the dataset. The reason for this is likely to be differences in the completeness of the known positive sets. For all five datasets, the 'real' positive sets are unknown and the disparity between genome size and the number of known positives means that they are likely to represent a small proportion of the 'real' positive set. As a consequence, the AUC values are likely to underestimate the retrieval capability of the algorithm. This also suggests that the IPX may not be optimised. We anticipate that as coverage and categorisation of protein-protein interaction data becomes available it will be possible to optimise the IPX, improve these AUC values and the utility of the metric.

ROC and IPX score distributions. Mirrored ROC curves for the five genome pairs in the known positive sets in KP. Inset: IPX score distributions (reproduced in Additional File 3: 'Interolog Prioritisation Index Histograms'). For each characteristic, the point at coordinate (1,1) corresponds to IPXthr = min(IPX), TPR = 100% and FPR = 100%. The point at coordinate (0,0) corresponds to IPXthr = max(IPX), TPR = 0% and FPR = 0%. Initially, IPXthr = min(IPX). Then, the score histogram is divided into 1000 segments and IPXthr is incremented until IPXthr = max(IPX) is reached. For all datasets, the decrease of TPR is slower than the decrease of FPR as IPXthr → max(IPX). This means that, as the score threshold becomes more stringent, for all datasets the number of known positive samples lost stays smaller than the number of new predictions lost. The correlation between TPR and the FPR varies depending on the dataset: in the case of the Yeast-Worm pair, 98% of known positives are retrieved when the novel prediction retrieval rate is down to about 76%. Conversely, in the Human-Yeast case, the TPR is down to about 92% for 98% FPR.
The reason why a number of known positives have a low index lies in the nature of the IPX. It is designed to reward functionally conserved interologs obtained from binary experimental PPIs. As stated above, the IPX penalises putative PPIs that are from orthology projections where co-orthologues exist or from binary interactions that have been artificially extracted from protein complexes. Some known positives will fall into one or both of these two categories. Additional File 3: 'Interolog Prioritisation Index Histograms' shows IPX distributions for the five known positive datasets in KP. Additionally, we show the distribution of the known positives within the IPX histograms in Additional File 4: 'Distribution of positive samples within the IPX histograms'. This chart shows, for each dataset, how many positive samples are in the low (dark), average (medium) and high (bright) tiers of the IPX distribution. For all but the mouse-human genome pair, most known positives fall in the second tier, and the mouse-human dataset is the only one to have most of its positives in the high tier. We examined the relationship between the IPX and the loss of known positives for the five sample datasets (Figure 6 and Additional File 5: 'TPR, FPR and IPX Threshold'). The mouse-human dataset preserves 80% of the positives with an IPXthr = 15. At the same threshold value, all of the other datasets fare significantly worse (Fisher Exact Test, Additional File 2: 'Definitions', Tables S2 and S3). These results reflect the closer phylogenetic distance between mouse and human, in which less gene duplication will have occurred since divergence from their common ancestor in comparison to the other species pairs.

TPR/IPX Threshold Curve. Relationship between IPX threshold and known positive sample loss for the five sample datasets. The mouse-human dataset preserves 80% of the positives with a IPXthr = 15. At the same threshold value, all the other studied datasets fare significantly worse (Fisher Exact Test, Additional File 2: 'Definitions', Tables S2 and S3).
It is important to stress that the IPX is not a necessary and sufficient condition to assess the reliability of a putative PPI: a highly ranking interolog may not turn out to be an interesting candidate, but as the index is calculated using both experimental and phylogenetic measures, it would be logical to assess such interologs as candidates before those with lower values. As is the case with any biological scoring metric, a feature with a low score may turn out to be a good candidate and should not be excluded solely on the basis of the score alone.
Example — Exploring the protein interactome of Drosophila melanogaster
To demonstrate the use of Bio::Homology::InterologWalk, we retrieved a list of all Drosophila melanogaster genes (DS_DMEL) from Ensembl-Compara Release 61. For the interolog walk the reference genomes were not restricted to any specific species, but included all 53 available taxa even though we expected a proportion of the species in the sets to provide 'dead end' orthologues where no significant experimental PPI data existed.
For the interolog walk we filtered by retrieving only one-to-one orthologues in the orthology mapping phases and discarded (a) all interactions that were inferred from complexes (spoke) and (b) all interactions that were not experimental physical associations. As a reference we also performed a direct mine of known interactions for the starting gene list with the same interactions filters.
Table 1 shows statistics for the resulting datasets. We adopt the following terminology:
-
1.
NET_DS_DMEL_known (7779 nodes, 26622 edges) - the network consisting of all the experimentally-obtained physical associations involving genes in DS_DMEL, according to EBI-Intact;
-
2.
NET_DS_DMEL_putative (2188 nodes, 4428 edges) - the network consisting of putative interactions involving genes in DS_DMEL according to Bio::Homology::InterologWalk (filtered as described above);
-
3.
NET_DS_DMEL_union (8270 nodes, 31050 edges) - the network obtained computing the union of (1) and (2) where:
-
each node is a node of NET_DS_DMEL_known, NET_DS_DMEL_putative, or both;
-
each edge is either an edge of NET_DS_DMEL_known or an edge of NET_DS_DMEL_putative (Note: duplicate edges were not collapsed into one).
In order to explore the results of the interolog walk we analysed the networks using the network tool Cytoscape [39]. Due to the size and complexity of the genome scale interaction networks we decided for the purposes of this illustration to focus on the 65 nodes in NET_DS_DMEL_known that were annotated with the term 'DNA replication' in the Gene Ontology [55]. In order to allow clear visualisation of the data, we further restricted this to a subset of 10 randomly selected genes. We then retrieved all of their nearest neighbours in NET_DS_DMEL_known which produced five disconnected networks (Figure 7A, 46 nodes and 53 edges in total) the biggest of which features 4 DNA replication genes (Figure 7A-1).

'DNA Replication' sub-network inNET_DS_DMEL_knownandNET_DS_DMEL_union. A: Data extracted from NET_DS_DMEL_known as follows: 1. select 10 core genes annotated with DNA replication GO biological process. 2. select all their nearest neighbours. B: NET_DNArep. Obtained as before, but data are extracted from from NET_DS_DMEL_union. B-D: Using the IPX to refine the putative sub-network in B. We set  (C) and
(C) and  (D). IPX score values are mapped to edge thickness in B-D. Figure 7D shows the sub-network backbone possessing the highest confidence according to the algorithm. Black nodes are genes annotated with the DNA Replication GO biological process. Black connections are experimental PPI data from EBI Intact while red connections are putative predictions taken from NET_DS_DMEL_putative. Nodes are described in key. A high-resolution version of B is presented in Additional File 6: 'NET_DNARep'.
(D). IPX score values are mapped to edge thickness in B-D. Figure 7D shows the sub-network backbone possessing the highest confidence according to the algorithm. Black nodes are genes annotated with the DNA Replication GO biological process. Black connections are experimental PPI data from EBI Intact while red connections are putative predictions taken from NET_DS_DMEL_putative. Nodes are described in key. A high-resolution version of B is presented in Additional File 6: 'NET_DNARep'.
To illustrate the utility of the interolog walk we performed the same procedure as above using NET_DS_DMEL_union. In this case we retrieved a set of 68 'DNA replication' genes, a superset of the 65 found before, meaning that 3 additional DNA replication genes are drawn in through the putative pipeline. As before, we selected the sub-network of NET_DS_DMEL_union composed of the 10 core DNA replication genes and their nearest neighbours. The resulting sub-network, NET_DNArep, composed of 68 nodes and 165 edges (Figure 7B and for clarity in higher resolution see Additional File 6: 'NET_DNArep') has greatly increased connectivity (compare Figure 7A to 7B). Indeed the main connected component in NET_DNArep now comprises 55 genes and 153 interactions, and wires together 7 of the 10 core DNA replication genes. A broad analysis of NET_DNArep reveals that the introduction of putative PPI data has allowed us to:
-
1.
collect evidence about new genes, not known to be related to DNA replication before;
-
2.
increase the connectivity of the GO-annotated DNA replication proteins.
Proteins that were known to be involved in DNA replication now interact with proteins for which no evidence for DNA replication involvement existed, meaning new potential candidates are drawn in to build a more complete picture of the domain.
Using the IPX to refine the Sub-Network
Given the relatively high number of interactions and participating genes in NET_DNArep, we carried out a refinement of the interaction candidates obtained, using the IPX. As described earlier, Bio::Homology::InterologWalk can optionally calculate a prioritisation index for each of the putative PPIs produced. These can be employed to define a 'core' network for which there is strong biological and experimental evidence by removing nodes that are connected by putative PPIs with low IPX indices.
In order to look at the composition of putative PPIs in NET_DNArep, we set  ,
,  and mapped score values to edge thickness in NET_DNArep (Figure 7B-D). Figure 7D shows the sub-network backbone possessing the highest confidence according to the algorithm. Interestingly, the connectedness of the main component still remains at this threshold level. Four putative PPIs survive the strict threshold: (Orc2, Mcm10), (MCM5, MCM3), (MCM10, MCM6) and (crn, CG6905). All these pairs, apart from (MCM6, MCM10), are known to interact experimentally (black edge) and the putative interaction (red edge) reconfirms these results. The (MCM6, MCM10) pair, on the other hand, has not been shown to interact in Drosophila melanogaster and represents a candidate for lab validation.
and mapped score values to edge thickness in NET_DNArep (Figure 7B-D). Figure 7D shows the sub-network backbone possessing the highest confidence according to the algorithm. Interestingly, the connectedness of the main component still remains at this threshold level. Four putative PPIs survive the strict threshold: (Orc2, Mcm10), (MCM5, MCM3), (MCM10, MCM6) and (crn, CG6905). All these pairs, apart from (MCM6, MCM10), are known to interact experimentally (black edge) and the putative interaction (red edge) reconfirms these results. The (MCM6, MCM10) pair, on the other hand, has not been shown to interact in Drosophila melanogaster and represents a candidate for lab validation.
Conclusions
In this paper, we present Bio::Homology::InterologWalk, a Perl module to retrieve, prioritise and visualise putative Protein-Protein Interactions using interolog mapping. Unlike previous efforts, this Perl library (a) automatically connects to orthology/PPI data web-services to generate up-to-date predictions 'on the fly'. (b) outputs its predictions in the form of simple text files, allowing to use its methods, or the data it produces, within the context of pipeline-based work flows of wider scope (c) optionally flags the predictions on the basis of related biological metadata through a prioritisation index, allowing the selection of a subset of candidates for in vivo validation.
We formally validate the efficacy of the tool and present ROC curves to assess the association between the IPX and 'true-positive' interactions across several inter-species 'true-positive' sets. We test the potential of the tool to retrieve putative PPIs on the Drosophila melanogaster genome and look more closely at one DNA replication related sub-network identifying several novel nodes and interactions. We conclude by using IPX thresholds to create a 'core' network for which there is strong biological and experimental support.
Our interaction prioritisation index (IPX) is designed to encapsulate biologically relevant principles that relate directly to the assessments currently made manually by many researchers using interaction data. We stress, however, that the IPX measure for an interaction is not fully explored here and that a full validation is not possible due to the current poor coverage of PPI data across species. In our experience the IPX has proven to be a useful summary of biological metadata for PPIs. When performing an interolog walk we recommend the user first uses filtering options to restrict the interactions retrieved and then uses the IPX as a pragmatic aid to candidate prioritisation.
Bio::Homology::InterologWalk is freely available for non-commercial purposes on the Comprehensive Perl Archive Network (CPAN) and modifiable under the GNU GPL license. The package includes full documentation and example scripts to simplify usage.
Availability and requirements
Project name : Bio::Homology::InterologWalk
Project home page : http://search.cpan.org/~ggallone/Bio-Homology-InterologWalk/
Programming Language : Perl
Other requirements : Bioperl, Ensembl API. The module also relies on a number of pre-requisite Perl libraries. See manual on website.
License : GNU GPL
Abbreviations
- API :
-
Application Programming Interface
- COG :
-
Cluster Of Orthologs
- CPAN :
-
Comprehensive Perl Archive Network
- EBI :
-
European Bioinformatics Institute
- IPX :
-
Interolog Prioritisation indeX
- GO :
-
Gene Ontology
- HUPO-PSI :
-
HUman Proteome Organisation - Proteomics Standards Initiative
- MITAB :
-
Molecular Interactions TAB delimited data exchange format
- PCS :
-
PPI Conservation Score
- PPI :
-
Protein-Protein Interaction
- PSICQUIC :
-
Protemics Standard Initiative Common QUery InterfaCe
- PSI-MI :
-
Proteomics Standard Initiative - Molecular Interactions
- REST :
-
Representational State Transfer
- ROC :
-
Receiver Operating Characteristic
- TAP :
-
Tandem Affinity Purification
- TreeBeST :
-
(gene) Tree Building guided by Species Tree.
References
Bray D: Molecular Networks: The Top-Down View. Science 2003, 301(5641):1864–1865. [http://www.sciencemag.org/cgi/content/abstract/301/5641/1864] 10.1126/science.1089118
Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, Vijayadamodar G, Pochart P, Machineni H, Welsh M, Kong Y, Zerhusen B, Malcolm R, Varrone Z, Collis A, Minto M, Burgess S, McDaniel L, Stimpson E, Spriggs F, Williams J, Neurath K, Ioime N, Agee M, Voss E, Furtak K, Renzulli R, Aanensen N, Carrolla S, Bickelhaupt E, Lazovatsky Y, DaSilva A, Zhong J, Stanyon CA, Finley JRL, White KP, Braverman M, Jarvie T, Gold S, Leach M, Knight J, Shimkets RA, McKenna MP, Chant J, Rothberg JM: A Protein Interaction Map of Drosophila melanogaster. Science 2003, 302(5651):1727–1736. [http://www.sciencemag.org/cgi/content/abstract/302/5651/1727] 10.1126/science.1090289
Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JDJ, Chesneau A, Hao T, Goldberg DS, Li N, Martinez M, Rual JF, Lamesch P, Xu L, Tewari M, Wong SL, Zhang LV, Berriz GF, Jacotot L, Vaglio P, Reboul J, Hirozane-Kishikawa T, Li Q, Gabel HW, Elewa A, Baumgartner B, Rose DJ, Yu H, Bosak S, Sequerra R, Fraser A, Mango SE, Saxton WM, Strome S, van den Heuvel S, Piano F, Vandenhaute J, Sardet C, Gerstein M, Doucette-Stamm L, Gunsalus KC, Harper JW, Cusick ME, Roth FP, Hill DE, Vidal M: A Map of the Interactome Network of the Metazoan C. elegans. Science 2004, 303(5657):540–543. [http://www.sciencemag.org/cgi/content/abstract/303/5657/540] 10.1126/science.1091403
LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE: A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 2005, 438(7064):103–107. 10.1038/nature04104
Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksöz E, Droege A, Krobitsch S, Korn B, Birchmeier W, Lehrach H, Wanker EE: A Human Protein-Protein Interaction Network: A Resource for Annotating the Proteome. Cell 2005, 122(6):957–968. [http://www.sciencedirect.com/science/article/B6WSN-4H3YGBS-1/2/d39ee6e848fc3d640dcccc8f9ce59eaf] 10.1016/j.cell.2005.08.029
Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual JF, Dricot A, Vazquez A, Murray RR, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet AS, Motyl A, Hudson ME, Park J, Xin X, Cusick ME, Moore T, Boone C, Snyder M, Roth FP, Barabasi AL, Tavernier J, Hill DE, Vidal M: High-Quality Binary Protein Interaction Map of the Yeast Interactome Network. Science 2008, 322(5898):104–110. [http://www.sciencemag.org/cgi/content/abstract/322/5898/104] 10.1126/science.1158684
Figeys D, McBroom LD, Moran MF: Mass Spectrometry for the Study of Protein-Protein Interactions. Methods 2001, 24(3):230–239. [http://www.sciencedirect.com/science/article/B6WN5–456JRHJ-18/2/6dc34bdb705478365b17cb5362d13b56] 10.1006/meth.2001.1184
Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrín-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MHY, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O'Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF: Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 2006, 440(7084):637–643. 10.1038/nature04670
Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O'Connor L, Li M, Taylor R, Dharsee M, Ho Y, Heilbut A, Moore L, Zhang S, Ornatsky O, Bukhman YV, Ethier M, Sheng Y, Vasilescu J, Abu-Farha M, Lambert JP, Duewel HS, Stewart II, Kuehl B, Hogue K, Colwill K, Gladwish K, Muskat B, Kinach R, Adams SL, Moran MF, Morin GB, Topaloglou T, Figeys D: Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol 2007., 3:
Valencia A, Pazos F: Computational methods for the prediction of protein interactions. Current Opinion in Structural Biology 2002, 12(3):368–373. [http://www.sciencedirect.com/science/article/B6VS6–469GK4C-J/2/9c30d085fb9e074bbf1d33355fd723f5] 10.1016/S0959-440X(02)00333-0
Berggård T, Linse S, James P: Methods for the detection and analysis of protein-protein interactions. Proteomics 2007, 7(16):2833–2842. 10.1002/pmic.200700131
Bork P, Dandekar T, Diaz-Lazcoz Y, Eisenhaber F, Huynen M, Yuan Y: Predicting function: from genes to genomes and back. Journal of Molecular Biology 1998, 283(4):707–725. [http://www.sciencedirect.com/science/article/B6WK7–45S492P-35/2/10490c37ccfb97ae7e78adebce867c5a] 10.1006/jmbi.1998.2144
Hegyi H, Gerstein M: Annotation Transfer for Genomics: Measuring Functional Divergence in Multi-Domain Proteins. Genome Research 2001, 11(10):1632–1640. [http://genome.cshlp.org/content/11/10/1632.abstract] 10.1101/gr. 183801
Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M: Protein Interaction Mapping in C. elegans Using Proteins Involved in Vulval Development. Science 2000, 287(5450):116–122. [http://www.sciencemag.org/cgi/content/abstract/287/5450/116] 10.1126/science.287.5450.116
Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, Garrels J, Vincent S, Vidal M: Identification of Potential Interaction Networks Using Sequence-Based Searches for Conserved Protein-Protein Interactions or "Interologs". Genome Research 2001, 11(12):2120–2126. [http://genome.cshlp.org/content/11/12/2120.abstract] 10.1101/gr.205301
Huang TW, Tien AC, Huang WS, Lee YCG, Peng CL, Tseng HH, Kao CY, Huang CYF: POINT: a database for the prediction of protein-protein interactions based on the orthologous interactome. Bioinformatics 2004, 20(17):3273–3276. [http://bioinformatics.oxfordjournals.org/content/20/17/3273.abstract] 10.1093/bioinformatics/bth366
Lehner B, Fraser A: A first-draft human protein-interaction map. Genome Biology 2004, 5(9):R63. [http://genomebiology.com/2004/5/9/R63] 10.1186/gb-2004-5-9-r63
Brown KR, Jurisica I: Online Predicted Human Interaction Database. Bioinformatics 2005, 21(9):2076–2082. [http://bioinformatics.oxfordjournals.org/content/21/9/2076.abstract] 10.1093/bioinformatics/bti273
Persico M, Ceol A, Gavrila C, Hoffmann R, Florio A, Cesareni G: HomoMINT: an inferred human network based on orthology mapping of protein interactions discovered in model organisms. BMC Bioinformatics 2005, 6(Suppl 4):S21. [http://www.biomedcentral.com/1471–2105/6/S4/S21] 10.1186/1471-2105-6-S4-S21
Kemmer D, Huang Y, Shah S, Lim J, Brumm J, Yuen M, Ling J, Xu T, Wasserman W, Ouellette BF: Ulysses - an application for the projection of molecular interactions across species. Genome Biology 2005, 6(12):R106. [http://genomebiology.com/2005/6/12/R106] 10.1186/gb-2005-6-12-r106
Gandhi TKB, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, Sharma S, Pinkert S, Nagaraju S, Periaswamy B, Mishra G, Nandakumar K, Shen B, Deshpande N, Nayak R, Sarker M, Boeke JD, Parmigiani G, Schultz J, Bader JS, Pandey A: Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 2006, 38(3):285–293. 10.1038/ng1747
Huang TW, Lin CY, Kao CY: Reconstruction of human protein interolog network using evolutionary conserved network. BMC Bioinformatics 2007, 8: 152. [http://www.biomedcentral.com/1471–2105/8/152] 10.1186/1471-2105-8-152
Wojcik J, Boneca IG, Legrain P: Prediction, Assessment and Validation of Protein Interaction Maps in Bacteria. Journal of Molecular Biology 2002, 323(4):763–770. [http://www.sciencedirect.com/science/article/B6WK7–473VN5W-F/2/786fdc31b9e4e8c7bc795c0f423c5859] 10.1016/S0022-2836(02)01009-4
Sharan R, Ideker T, Kelley B, Shamir R, Karp RM: Identification of Protein Complexes by Comparative Analysis of Yeast and Bacterial Protein Interaction Data. Journal of Computational Biology 2005, 12(6):835–846. [http://www.liebertonline.com/doi/abs/10.1089/cmb.2005.12.835] 10.1089/cmb.2005.12.835
Wuchty S, Ipsaro JJ: A Draft of Protein Interactions in the Malaria Parasite P. falciparum. Journal of Proteome Research 2007, 6(4):1461–1470. [http://pubs.acs.org/doi/abs/10.1021/pr0605769] 10.1021/pr0605769
He F, Zhang Y, Chen H, Zhang Z, Peng YL: The prediction of protein-protein interaction networks in rice blast fungus. BMC Genomics 2008, 9: 519. [http://www.biomedcentral.com/1471–2164/9/519] 10.1186/1471-2164-9-519
Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JDJ, Bertin N, Chung S, Vidal M, Gerstein M: Annotation Transfer Between Genomes: Protein-Protein Interologs and Protein-DNA Regulogs. Genome Research 2004, 14(6):1107–1118. [http://genome.cshlp.org/content/14/6/1107.abstract] 10.1101/gr.1774904
Michaut M, Kerrien S, Montecchi-Palazzi L, Chauvat F, Cassier-Chauvat C, Aude JC, Legrain P, Hermjakob H: InteroPORC: automated inference of highly conserved protein interaction networks. Bioinformatics 2008, 24(14):1625–1631. [http://bioinformatics.oxfordjournals.org/content/24/14/1625.abstract] 10.1093/bioinformatics/btn249
Wiles A, Doderer M, Ruan J, Gu TT, Ravi D, Blackman B, Bishop A: Building and analyzing protein interactome networks by cross-species comparisons. BMC Systems Biology 2010, 4: 36. [http://www.biomedcentral.com/1752–0509/4/36] 10.1186/1752-0509-4-36
Kersey P, Bower L, Morris L, Horne A, Petryszak R, Kanz C, Kanapin A, Das U, Michoud K, Phan I, Gattiker A, Kulikova T, Faruque N, Duggan K, Mclaren P, Reimholz B, Duret L, Penel S, Reuter I, Apweiler R: Integr8 and Genome Reviews: integrated views of complete genomes and proteomes. Nucleic Acids Research 2005, 33(suppl 1):D297-D302. [http://nar.oxfordjournals.org/content/33/suppl_1/D297.abstract]
Pedamallu CS, Posfai J: Open source tool for prediction of genome wide protein-protein interaction network based on ortholog information. Source Code for Biology and Medicine 2010, 5: 8. [http://www.scfbm.org/content/5/1/8] 10.1186/1751-0473-5-8
Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, Lehväslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, Birney E: The Bioperl Toolkit: Perl Modules for the Life Sciences. Genome Research 2002, 12(10):1611–1618. [http://genome.cshlp.org/content/12/10/1611.abstract] 10.1101/gr.361602
Kersey PJ, Lawson D, Birney E, Derwent PS, Haimel M, Herrero J, Keenan S, Kerhornou A, Koscielny G, Kähäri A, Kinsella RJ, Kulesha E, Maheswari U, Megy K, Nuhn M, Proctor G, Staines D, Valentin F, Vilella AJ, Yates A: Ensembl Genomes: Extending Ensembl across the taxonomic space. Nucleic Acids Research 2010, 38(suppl 1):D563-D569. [http://nar.oxfordjournals.org/content/38/suppl_1/D563.abstract]
Flicek P, Aken BL, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Coates G, Fairley S, Fitzgerald S, Fernandez-Banet J, Gordon L, Gräf S, Haider S, Hammond M, Howe K, Jenkinson A, Johnson N, Kähäri A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Koscielny G, Kulesha E, Lawson D, Longden I, Massingham T, McLaren W, Megy K, Overduin B, Pritchard B, Rios D, Ruffier M, Schuster M, Slater G, Smedley D, Spudich G, Tang YA, Trevanion S, Vilella A, Vogel J, White S, Wilder SP, Zadissa A, Birney E, Cunningham F, Dunham I, Durbin R, Fern_andez-Suarez XM, Herrero J, Hubbard TJP, Parker A, Proctor G, Smith J, Searle SMJ: Ensembl's 10th year. Nucleic Acids Research 2010, 38(suppl 1):D557-D562. [http://nar.oxfordjournals.org/content/38/suppl_1/D557.abstract]
Kerrien S, Orchard S, Montecchi-Palazzi L, Aranda B, Quinn A, Vinod N, Bader G, Xenarios I, Wojcik J, Sherman D, Tyers M, Salama J, Moore S, Ceol A, Chatr-aryamontri A, Oesterheld M, Stumpen V, Salwinski L, Nerothin J, Cerami E, Cusick M, Vidal M, Gilson M, Armstrong J, Woollard P, Hogue C, Eisenberg D, Cesareni G, Apweiler R, Hermjakob H: Broadening the horizon - level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biology 2007, 5: 44. [http://www.biomedcentral.com/1741–7007/5/44] 10.1186/1741-7007-5-44
Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K, Hermjakob H: The IntAct molecular interaction database in 2010. Nucleic Acids Research 2010, 38(suppl 1):D525-D531. [http://nar.oxfordjournals.org/content/38/suppl_1/D525.abstract]
The Comprehensive Perl Archive Network[http://www.cpan.org/]
Gallone G: Bio::Homology::InterologWalk - Retrieve, score and visualize putative Protein-Protein Interactions through the orthology-walk method.2011. [http://search.cpan.org/~ggallone/Bio-Homology-InterologWalk/]
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T: Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Research 2003, 13(11):2498–2504. [http://genome.cshlp.org/content/13/11/2498.abstract] 10.1101/gr.1239303
Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YHC, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, Pablos Bd, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RDC, Scheeler F, Shen H, Shue BC, Sidén-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC: The Genome Sequence of Drosophila melanogaster. Science 2000, 287(5461):2185–2195. [http://www.sciencemag.org/content/287/5461/2185.abstract] 10.1126/science.287.5461.2185
Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E: EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Research 2009, 19(2):327–335. [http://genome.cshlp.org/content/19/2/327.abstract]
Fielding RT, Taylor RN: Principled design of the modern Web architecture. ICSE '00: Proceedings of the 22nd international conference on Software engineering New York, NY, USA: ACM; 2000, 407–416. [http://portal.acm.org/citation.cfm?id=337228]
Prieto C, De Las Rivas J: APID: Agile Protein Interaction DataAnalyzer. Nucleic Acids Research 2006, 34(suppl 2):W298-W302. [http://nar.oxfordjournals.org/content/34/suppl_2/W298.abstract]
Razick S, Magklaras G, Donaldson I: iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinformatics 2008, 9: 405. [http://www.biomedcentral.com/1471–2105/9/405] 10.1186/1471-2105-9-405
Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, Oughtred R, Lackner DH, Bähler J, Wood V, Dolinski K, Tyers M: The BioGRID Interaction Database: 2008 update. Nucleic Acids Research 2008, 36(suppl 1):D637-D640. [http://nar.oxfordjournals.org/content/36/suppl_1/D637.abstract]
Goll J, Rajagopala SV, Shiau SC, Wu H, Lamb BT, Uetz P: MPIDB: the microbial protein interaction database. Bioinformatics 2008, 24(15):1743–1744. [http://bioinformatics.oxfordjournals.org/content/24/15/1743.abstract] 10.1093/bioinformatics/btn285
Chautard E, Ballut L, Thierry-Mieg N, Ricard-Blum S: MatrixDB, a database focused on extracellular protein-protein and protein-carbohydrate interactions. Bioinformatics 2009, 25(5):690–691. [http://bioinformatics.oxfordjournals.org/content/25/5/690.abstract] 10.1093/bioinformatics/btp025
Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C: STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research 2009, 37(suppl 1):D412-D416. [http://nar.oxfordjournals.org/content/37/suppl_1/D412.abstract]
Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, Kanapin A, Lewis S, Mahajan S, May B, Schmidt E, Vastrik I, Wu G, Birney E, Stein L, D'Eustachio P: Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Research 2009, 37(suppl 1):D619-D622. [http://nar.oxfordjournals.org/content/37/suppl_1/D619.abstract]
Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, Cesareni G: MINT, the molecular interaction database: 2009 update. Nucleic Acids Research 2010, 38(suppl 1):D532-D539. [http://nar.oxfordjournals.org/content/38/suppl_1/D532.abstract]
He X, Zhang J: Rapid Subfunctionalization Accompanied by Prolonged and Substantial Neofunctionalization in Duplicate Gene Evolution. Genetics 2005, 169(2):1157–1164. [http://www.genetics.org/cgi/content/abstract/169/2/1157] 10.1534/genetics.104.037051
Hittinger CT, Carroll SB: Gene duplication and the adaptive evolution of a classic genetic switch. Nature 2007, 449(7163):677–681. 10.1038/nature06151
Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW: Evolutionary Rate in the Protein Interaction Network. Science 2002, 296(5568):750–752. [http://www.sciencemag.org/cgi/content/abstract/296/5568/750] 10.1126/science.1068696
Wuchty S, Oltvai ZN, Barabasi AL: Evolutionary conservation of motif constituents in the yeast protein interaction network. Nat Genet 2003, 35(2):176–179. 10.1038/ng1242
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene Ontology: tool for the unification of biology. Nat Genet 2000, 25: 25–29. 10.1038/75556
Acknowledgements and Funding
Funding was provided by the Wellcome Trust (TIS, JDA, APJ) under grant number 077266, by the ERASysBio+ scheme (TIS) and by an MRC Capacity Building Studentship in Systems Biology (GG), number G68293X. This work has made use of resources provided by Ensembl http://www.ensembl.org and the EBI http://www.ebi.ac.uk. GG wishes to thank Lysimachos Zografos for interesting discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors' contributions
GG designed and wrote the Bio::Homology::InterologWalk module and performed all of the analyses. APJ, TIS and JDA supervised the work, and all authors contributed to the preparation of the final manuscript.
Electronic supplementary material
12859_2011_4720_MOESM1_ESM.PDF
Additional file 1:Simplified schematics of the Interolog Walk pipelines. Flow Diagram documenting the structure and data sources on which the Bio::Homology::InterologWalk pipeline implementation is based. (PDF 1013 KB)
12859_2011_4720_MOESM2_ESM.PDF
Additional file 2:Definitions. Supplementary text providing Bio::Homology::InterologWalk implementation details, design decisions and mathematical background. (PDF 214 KB)
12859_2011_4720_MOESM3_ESM.PDF
Additional file 3:Interolog Prioritisation Index Histograms. IPX Histograms for the five putative PPI datasets built from the Positive datasets. (PDF 65 KB)
12859_2011_4720_MOESM4_ESM.PDF
Additional file 4:Distribution of positive samples within the IPX histograms. Distribution of known positive samples in the IPX histograms. The chart shows, for each of the datasets in KP, the number of known positive samples in the low (dark), average (medium) and high (bright) tiers of the IPX distribution. (PDF 37 KB)
12859_2011_4720_MOESM5_ESM.PDF
Additional file 5:TPR, FPR and IPX Threshold. Relationship between TPR, FPR and IPX Threshold for the five putative PPI datasets obtained from the Positive datasets through Bio::Homology::InterologWalk. (PDF 53 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gallone, G., Simpson, T.I., Armstrong, J.D. et al. Bio::Homology::InterologWalk - A Perl module to build putative protein-protein interaction networks through interolog mapping. BMC Bioinformatics 12, 289 (2011). https://doi.org/10.1186/1471-2105-12-289
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-12-289