- Methodology article
- Open access
- Published:
HMM Logos for visualization of protein families
BMC Bioinformatics volume 5, Article number: 7 (2004)
Abstract
Background
Profile Hidden Markov Models (pHMMs) are a widely used tool for protein family research. Up to now, however, there exists no method to visualize all of their central aspects graphically in an intuitively understandable way.
Results
We present a visualization method that incorporates both emission and transition probabilities of the pHMM, thus extending sequence logos introduced by Schneider and Stephens. For each emitting state of the pHMM, we display a stack of letters. The stack height is determined by the deviation of the position's letter emission frequencies from the background frequencies. The stack width visualizes both the probability of reaching the state (the hitting probability) and the expected number of letters the state emits during a pass through the model (the state's expected contribution).
A web interface offering online creation of HMM Logos and the corresponding source code can be found at the Logos web server of the Max Planck Institute for Molecular Genetics http://logos.molgen.mpg.de.
Conclusions
We demonstrate that HMM Logos can be a useful tool for the biologist: We use them to highlight differences between two homologous subfamilies of GTPases, Rab and Ras, and we show that they are able to indicate structural elements of Ras.
Background
Introduction
Many existing gene or protein sequences in different organisms are related through evolution and can be grouped into families. One way of representing such a family is through a profile Hidden Markov Model (pHMM). A pHMM is a fully probabilistic generative model; it specifies position-specific letter emission distributions and also position-specific insertion and deletion probabilities to describe a sequence family. The existence of efficient algorithms for pHMM creation and database search [1] makes pHMMs the tool of choice for protein family research. For example, the protein family and domain databases Pfam [2] and SMART [3] both use pHMMs. However, the large number of parameters in the underlying model makes it non-trivial to present a visual overview of the characteristics that make up a family.
For sequence profiles, also known as position-specific score matrices (PSSMs), and ungapped multiple alignments, there exists a visualization method called the Sequence Logo [4]. A Sequence Logo graphically represents the conservation of the columns (positions) in a multiple alignment by plotting a stack of letters (nucleotides or amino acids) for each position. The total stack height is computed as the information content of the column, i.e., its relative entropy distance from an assumed background distribution. The relative height of each letter in the stack is proportional to its frequency at the position. Usually, colors are used to represent different properties of the letters (e.g., green for aromatic amino acids).
If we ignore the position-specific insertion and deletion probabilities of a pHMM, we can treat is as a PSSM and visualize it with a sequence logo (the makelogo tool of the SAM software package [5] does exactly this), but this would mean throwing away a substantial part of the available information. Therefore our aim is to modify Sequence Logos in such a way that they give an impression of the central aspects of pHMMs: Which positions can be deleted, which ones are highly conserved, and where can we expect long insertions?
Profiles and sequence logos
Let Σ be an alphabet and |Σ| its cardinality. For DNA, |Σ| = 4, and the letters of the alphabet are the four nucleotides A, C, G, and T. For proteins, |Σ| = 20, and the letters are the twenty amino acids.
A profile is a probabilistic description of a sequence. It specifies a probability distribution over the alphabet's letters for each position. More formally, a profile P of length L over Σ is an L x |Σ| matrix (P ij ) (i = 1,...,L; j ∈ Σ), such that P ij ≥ 0 for all i, j and Σj∈ΣP ij = 1 for all i.
A multiple sequence alignment of N sequences with L columns or positions can be interpreted as a profile. Let C ij be the number of occurrences of letter j ∈ Σ at position i, and let N i ≔ Σj ∈ ΣC ij ≤ N be the number of non-gap letters at position i. Then the maximum likelihood (ML) estimation of the profile P associated with this alignment is given by P ij ≔ C ij /N i . When the multiple alignment contains only few sequences, ML estimation results in many "impossibilities" (zero probabilities) in the profile and hence in over-fitting the model to the small sample. To counteract this problem, the profile is regularized, either by using Dirichlet mixture priors [6], or by alternative techniques (e.g., [7]).
The uncertainty or entropy [8] of distribution P i at the i-th position of the profile is given by H(P i ) = -Σj ∈ ΣP ij log2 P ij . The entropy H(P i ) is always nonnegative. It vanishes if and only if P i is a Dirac distribution, i.e., if the whole mass is accumulated at a single letter. The entropy takes its maximal value of log2 |Σ| bits (2 bits for DNA, approximately 4.32 bits for proteins) when P i is the uniform distribution, i.e., when P ij = 1/|Σ| for all j [9]. Since we use the binary logarithm log2, the unit of the entropy is called a "bit". When we use the natural logarithm, it is called a "nat", and for log10, it is called a "dit".
We may define the information content I(P i ) of position i as the "opposite" of its uncertainty,

The information content is a number between 0 and log2 |Σ| bits and measures the conservation of a position in a profile.
Since conserved positions in sequence families are considered to be functionally or structurally important, they should stand out when the profile is visualized. Schneider and Stephens [4] achieved this goal by representing each position by a stack of letters, where the stack height at position i is precisely the information content I (P i ).
While this method works well on DNA alignments, additional considerations must be made for protein sequences. Amino acids naturally occur with different "background" frequencies. For example, tryptophan (W) occurs much less frequently than leucine (L). The background frequencies might be computed by counting amino acid occurrences in all known proteins, or only in the proteins of the superfamily under consideration. Assume that the background frequency of amino acid j is π j > 0. Then the important positions are those whose distribution differs from π. Therefore it has become common practice to consider the relative entropy between the distributions P i and π,

where 0·log2(0/π j ) ≔ 0 by continuity as long as π j > 0.
Note that for the uniform distribution π j = 1/|Σ|, we have H(P i || π) = I(P i ). Thus the information content of P i , as defined above, is its relative entropy distance from the uniform distribution.
In a classical Sequence Logo, the stack height at position i is H(P i || π), the height of letter j within the stack is P ij H(P i || π), the letters are stacked in sorted order, the largest letter being on top of the stack, and colors may be used to highlight different properties of different letters. The HMM Logo inherits all of these characteristics, but also has additional ones to represent the additional information contained in a pHMM.
Profile HMMs
An HMM is a discrete time Markov chain that emits a letter from the alphabet Σ whenever a state is visited. The central idea is that only the emitted letters can be observed, but that the state sequence is hidden. A comprehensive review on the topic of HMMs can be found in the literature [10]. Profile HMMs are a specialization of HMMs to represent sequence families. Applications to protein modeling were first described in a paper by Krogh and co-workers [11], and are reviewed, for example, by Eddy [1].
Figure 1 shows the transition graph of a pHMM according to the HMMER software package [12]. For each position i (a consensus column of the underlying multiple alignment), a "match" state M
i
models the distribution E
i
of emitted letters at that position; it corresponds exactly to the profile distribution P
i
. An "insert" state I
i
allows for insertion of one or more letters between positions i and i + 1; their distribution 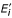 is individually specified for each insert state. A "delete" state D
i
is non-emitting and allows to pass the corresponding match state M
i
, resulting in a deletion at the i-th alignment position. The part consisting of the M
i
, I
i
, and D
i
states, flanked by the B and E states, is called the main model. There are further special states (S, N, J, C, and T) in Figure 1, which are not relevant for HMM Logos. As an exception, the background frequencies π of the letters may be learned from the emission probabilities of the N or C state, which represent flanking sequence.
is individually specified for each insert state. A "delete" state D
i
is non-emitting and allows to pass the corresponding match state M
i
, resulting in a deletion at the i-th alignment position. The part consisting of the M
i
, I
i
, and D
i
states, flanked by the B and E states, is called the main model. There are further special states (S, N, J, C, and T) in Figure 1, which are not relevant for HMM Logos. As an exception, the background frequencies π of the letters may be learned from the emission probabilities of the N or C state, which represent flanking sequence.

A path through the main model starts in the silent (non-emitting) begin state B, ends in E, and follows the legal state transitions. As in every Markov chain, state transitions have probabilities associated with them. We write As,tfor the transition probability s → t, so we have As,t≥ 0, Σ t As,t= 1 for all s, and As,t= 0 whenever no arrow s → t exists. There are exactly seven outgoing transitions (hence the model name Plan7) from every position i (except the last one): M i → I i , M i → Mi+1, M i → Di+1; D i → Mi+1, D i → Di+1; I i → Mi+1, and the self-loop I i → I i .
There are two major pHMM software packages, HMMER [12] and SAM [5], with small differences between their model topologies. So far we have described the HMMER model. The SAM model allows more state transitions: In addition to the transitions marked by the solid arrows in Figure 1, the transitions marked by dashed arrows, D i → I i and I i → Di+1are possible.
Results
HMM Logo concepts
The relevant information contained in a pHMM of length L can be summarized as
-
letter background frequencies π = (π j ), j ∈ Σ,
-
emission probabilities E = (E ij ) for match states (M i ), i = 1,...,L, j ∈ Σ,
-
emission probabilities E' = (
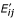 ) for insert states (I
i
), i = 1,...,L - 1, j ∈ Σ,
) for insert states (I
i
), i = 1,...,L - 1, j ∈ Σ, -
state transition probabilities A = (As,t).
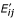 ) for insert states (I
i
), i = 1,...,L - 1, j
) for insert states (I
i
), i = 1,...,L - 1, j Sequence Logos can already take care of visualizing the emission probabilities in comparison to the background frequencies. We shall use the remaining dimension of a stack, its width, to visualize the transition probabilities in a meaningful way.
Each path B → … → E through the main model visits ("hits") certain states and misses others. For example, a path may hit either M i or D i , but not both. When a path hits an insert state I i , several letters may be emitted before it moves on to Mi+1. This leads to the following definitions.
Definition 1 (Hitting probability). Let s be a state of the main model. The hitting probability h(s) is the probability that a path B → … → E following the transition probabilities A, hits s at some point between B and E.
Definition 2 (Contribution). Let s be a match or insert state of the main model. Its contribution C(s) to an emitted sequence is a random variable describing the number of emitted letters in s along a path B → … → E. Further, we define c(s) ≔  [C(s)] as the expected contribution of state s.
[C(s)] as the expected contribution of state s.
Computation of hitting probabilities
The hitting probability of a state equals the sum of probabilities of all paths B → … → E visiting this state. Because of the self loops in insert states, this is an infinite number of paths. The hitting probability can nevertheless be computed efficiently using a forward-type dynamic programming algorithm as follows.
Proposition 1. Define  as the conditional probability that a path hitting Ii-1exits into M
i
. Then 1 - μ
i
is the probability of exiting into D
i
. For the Plan7 model disallowing the Ii-1→ D
i
transition we have μ
i
= 1. For the general SAM-type pHMM model allowing all 9 transitions, the hitting probabilities are
as the conditional probability that a path hitting Ii-1exits into M
i
. Then 1 - μ
i
is the probability of exiting into D
i
. For the Plan7 model disallowing the Ii-1→ D
i
transition we have μ
i
= 1. For the general SAM-type pHMM model allowing all 9 transitions, the hitting probabilities are
-
at the first position given by

-
at the following positions i ≥ 2 given by

Proof. The initializations for h(M1) and h(I1) are obvious from Figure 1. At every position i ≥ 1 we have h(D i ) = 1 - h(M i ) because each path passes either through Mi-1or Di-1.
For h(M i ), i ≥ 2, there are three ways into M i . The first term accounts for paths that come directly from Mi-1, the second term similarly accounts for direct entries from Di-1, and the last term accounts for paths that enter via Ii-1. A similar argument applies to the insert state hitting probabilities, for which there are only two ways of entry. All probabilities can be expressed solely in terms of h(Ii-1) as shown. □
Computation of expected contributions
The expected contribution of each state is easily derived from its hitting probability. Since delete states are non-emitting, their contribution is zero.
Proposition 2 (Expected contribution). We have
-
c(M i ) = h(M i ),
-
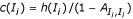
.
Proof. If a match state M i is hit, it contributes C = 1 letter; otherwise, it contributes nothing. This results in an expectation of c(M i ) = 1·h(M i ) + 0·(1 - h(M i )) = h(M i ).
If an insert state I
i
is hit, its contribution has a geometric distribution with "success parameter" (probability of leaving the state)  . Then the expected sojourn time is the reciprocal of this probability. If the state is not hit, its contribution is zero. Together, this results in an expectation of
. Then the expected sojourn time is the reciprocal of this probability. If the state is not hit, its contribution is zero. Together, this results in an expectation of 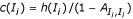 . □
. □
Proposition 3 (Expected number of emitted letters). The expected number of emitted letters during a walk from B to E through a profile HMM with L positions is

Proof. Every emitting state s emits on average c(s) letters during a walk from state B to E. The proposition follows from the linearity of expectation. □
We find it logical to set the width of the stack of an emitting state s to c(s), for then by Proposition 3, the total width of the logo represents the total number of emitted letters. However, we would like to display both the hitting probability and the expected contribution of a state. This is a non-problem for match states. For insert states s, we always have h(s) ≤ c(s), and so we can use two different background shadings for h(s) and for the remainder c(s) - h(s).
HMM Logo layout
The final definition of an HMM logo is as follows; see Figure 2 for a typical example.

Partial logo (positions 172–209) of the Pfam pkinase model. Positions with narrow match state stacks are likely to be deleted in typical family members. The total width of a red-shaded (dark+light) stack visualizes the expected number of inserted letters. The left dark-shaded part of the stack's width represents the probability that at least one letter is inserted. The difference is illustrated by comparing I173 with I176: Both states have approximately the same expected contribution, but the hitting probability of I176 is higher. The insertion stack height is zero for all shown examples because the emission probabilities correspond to the background frequencies.
-
HMM Logos consist of alternating stacks for match and insert states for all positions 1,...,L in the profile; the stack order is M1, I1, M2, I2,...,IL-1, M L .
-
The total height of a stack is the relative entropy H(e||π) between the state's emission distribution e and the background distribution π obtained from state N.
-
The relative height of letter j ∈ Σ within the stack is proportional to its emission probability e j .
-
The letters are stacked in sorted order, the largest letter being on top of the stack.
-
The total width of a stack s is its expected contribution c(s).
-
The background of an insert state's stack is shaded in two different colors for a total width of c(s) "letter units". The first h(s) units represent the hitting probability and are shaded with a medium-red background. The remaining c(s) - h(s) units are shaded with a lighter red.
-
The upper left corner of the logo shows a horizontal bar representing a contribution of 1 letter.
-
Insert state stacks are always displayed with a width of at least one pixel, thus making consecutive positions easier to distinguish.
-
Letters are drawn in different colors. The color scheme depends on the alphabet; amino acids are colored to represent structural or functional similarity.
-
The position number is displayed on the x-axis below every match/insert pair. The height of the y-axis is min(2, max i {H(E i || π), H(
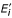 || π)}) bits, i.e., at least 2 bits, even if all stacks have a lower height.
|| π)}) bits, i.e., at least 2 bits, even if all stacks have a lower height.
Visualization of subfamily-specific sites
Since profile HMMs are predominantly used for protein family and domain modeling, we present examples that illustrate the utility of HMM Logos in this area.
While building an HMM for a domain, one usually tries to cover all homologous sequences. But, with ongoing experimental characterization, it often becomes clear that a single domain family consists of multiple, functionally divergent subfamilies.
Identifying these subfamilies and characterizing their determinants is an important step in protein function prediction. Creation of subfamily-specific profile HMMs is a first step in this direction performed by domain databases like SMART [3]. HMM Logos highlight regions which distinguish homologous subfamilies from each other and thereby facilitate the detection of subfamily determinants. Here, we illustrate this application on two subfamilies, Ras and Rab, of the small GTPases, whose profile HMMs were obtained from the SMART multiple subfamily alignment.
Combining sequence and structure analysis, Pereira-Leal and Seabra identified five regions which distinguish the Rab proteins from Ras like members [13]. Figure 3 depicts four of these regions (RabF2 to RabF5), which, in the three-dimensional structure, cluster between sheets β3 and β4. These are included in the switch II region, which changes conformation upon binding of GTP or GDP and mediates interactions with effectors and regulators. Furthermore, this region allows interactors to distinguish between Ras and Rab proteins and thus should contain subfamily determinants. By comparing the HMM logos for the two subfamilies, indeed both, domain and subfamily specific sites become apparent. For example, N-terminal to the small GTPase typical sequence DTAG, there is a highly conserved W in the Rab subfamily, whereas the corresponding site in Ras protein shows less conservation but a prevalence of the hydrophobic amino acids L or V.

Highlighting of loop regions
An important feature distinguishing HMM Logos from standard Sequence Logos is their ability to visualize regions with long expected inserts. These insertions usually do not happen within conserved structural elements, that is alpha helices or beta sheets, as this would influence and possibly break the structure of the whole domain. Instead, insertions are more likely to occur within loop regions.
Therefore the presence of frequent insertions at a given site can indicate that the site itself and its neighbors lie within a loop region. Figure 4 illustrates that this concept holds true for the HMM of the Ras domain. Here two regions with a prominent insert state can be found. Mapping them onto the known secondary structure (Protein Data Bank identifier PDB:121P) shows that these insert states indeed fall between the known structural elements.

Mapping of structural elements to a region of the Ras family HMM Logo. The mapping was obtained by aligning the sequence of p21 ras, the structure of which has been solved, to the Ras family pHMM. Below the logo, insert regions are highlighted by vertical arrows, and the secondary structure of p21 ras is indicated (alpha helices: barrels; beta sheets: horizontal arrows).
Discussion
The examples in the previous section illustrate the potential utility of HMM Logos, but they also point out a particularity of the HMMER software: In all Pfam and SMART pHMMs we looked at, the stack height in all insertion states is zero. This seems to be a consequence of HMMER's hmmbuild program: Insert states receive a very high emission prior that is equal to the background. This makes sense to allow the insertion of variable sequence parts of varying lengths at a position, i.e., in an insert state with high expected contribution. In order to change the emission probabilities away from the background, one would have to observe a consistent insertion that is common to several family members at the same position. Then however, hmmbuild would model this conserved "insertion" as a match state and model the sequences skipping this position via the delete state, even if this is the majority of the family members. This is immediately obvious from the numerous narrow match states shown in Figure 2. In our opinion, these narrow match states could be modeled more meaningfully as insert states with non-trivial emission probabilities. So while HMMER supports insert-specific emission probabilities, they do not seem to be used. HMM Logos immediately made this particularity visible; we were not aware of it before.
While we hope that HMM Logos can help to compare families visually, the RAS-RAB example (Figure 3) leaves us asking for more functionality: It would be useful to align two or several logos. In this way, a multiple family alignment of many sequences from a few different subfamilies could be represented as a multiple alignment of a few logos. Finding the most natural definition of the alignment score and the graphical representation of such a Logo alignment seem to be interesting topics for the future.
Conclusion
We have developed a method to visualize profile HMM specific information and demonstrated its utility for the biologist who wants to look at the model of a protein family or domain.
A PERL package for parsing and visualizing HMMER pHMMs is available under the GNU General Public License from the authors and can be downloaded from the Logos server of the Max Planck Institute for Molecular Genetics http://logos.molgen.mpg.de. At the same location we also offer the WWW-based tool LogoMat-M for HMM Logo generation which can be accessed in several ways. For example, in an interactive web form the user may specify a file in HMMER format which is uploaded and processed. Available options are described in the online help. Alternatively, it is possible to URL-encode Pfam identifiers, such as in
http://logos.molgen.mpg.de/cgi-bin/logomat-m.cgi?pfamid=AAA.
This will display a logo of the Pfam entry "ATPase family associated with various cellular activities" (AAA), using the default settings. Finally, the logos can be directly accessed from the Pfam website by pressing the "View HMM Logo" button on each domain's or family's overview page.
References
Eddy SR: Profile Hidden Markov Models. Bioinformatics 1998, 14: 755–63. 10.1093/bioinformatics/14.9.755
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res 2002, 30: 276–280. 10.1093/nar/30.1.276
Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, Ciccarelli F, Copley RR, Ponting CP, Bork P: Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res 2002, 30: 242–244. 10.1093/nar/30.1.242
Schneider TD, Stephens RM: Sequence Logos: A new way to display consensus sequences. Nucleic Acids Res 1990, 18: 6097–6100.
Hughey R, Karplus K, Krogh A: SAM Sequence Alignment and Modeling Software System. Technical Report UCSC-CRL-99–11, updated for SAM Version 3.4. Baskin School of Engineering University of California Santa Cruz 2003.
Sjølander K, Karplus K, Brown M, Hughey R, Krogh A, Mian IS, Haussler D: Dirichlet mixtures: a method for improved detection of weak but significant protein sequence homology. CABIOS 1996, 12(4):327–345.
Rahmann S, Müller T, Vingron M: On the Power of Profiles for Transcription Factor Binding Site Detection. Stat Appl Genet Mol Biol 2003, 2(1):Article 7. [http://www.bepress.com/sagmb/vol2/iss1/art7]
Shannon CE, Weaver W: The Mathematical Theory of Communication. Urbana: University of Illinois Press 1949.
Cover TM, Thomas JA: Elements of Information Theory. Wiley Series in Telecommunications John Wiley & Sons 1991.
Rabiner LR: A tutorial on Hidden Markov Models and selected applications in speech recognition. Proc IEEE 1989, 77: 257–286. 10.1109/5.18626
Krogh A, Brown M, Mian IS, Sjølander K, Haussler D: Hidden markov models in computational biology. Applications to protein modeling. J Mol Biol 1994, 235: 1501–31. 10.1006/jmbi.1994.1104
Eddy SR: HMMER User's Guide: Biological sequence analysis using profile Hidden Markov Models, version 2.2. Washington University School of Medicine 2001. [http://hmmer.wustl.edu]
Pereira-Leal JB, Seabra MC: The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol 2000, 301: 1077–1087. 10.1006/jmbi.2000.4010
Acknowledgements
We would like to express our gratitude to the PERL community; in particular to the creators of the PERL Data Language (PDL) and to the authors of the modules HMMERViewer (Robin Dowell), Imager (Arnar M. Hrafnkelsson and Tony Cook), and TFBS (Boris Lenhard). We thank Andrea Weiβe, Niels Köhler and Victoria Ornelas for valuable comments and discussions, David Studholme from the Sanger Center for information about direct Pfam access, Wilhelm Rüsing for help with the web server, and Martin Vingron for supervising the thesis of BSB. One of the anonymous referees provided valuable comments about applying the method to SAM-style HMMs.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SR had the initial idea to use the stack width to visualize the insertion and deletion probabilities. BSB implemented the software and the web server and invented the two-colored scheme for visualizing both hitting probability and expected contribution of an insert state. This work is part of his Bachelor's degree at the Free University of Berlin. JS examined the small GTPases with HMM Logos. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
About this article
Cite this article
Schuster-Böckler, B., Schultz, J. & Rahmann, S. HMM Logos for visualization of protein families. BMC Bioinformatics 5, 7 (2004). https://doi.org/10.1186/1471-2105-5-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-5-7