- Proceedings
- Open access
- Published:
Prediction of tissue-specific cis-regulatory modules using Bayesian networks and regression trees
BMC Bioinformatics volume 8, Article number: S2 (2007)
Abstract
Background
In vertebrates, a large part of gene transcriptional regulation is operated by cis-regulatory modules. These modules are believed to be regulating much of the tissue-specificity of gene expression.
Results
We develop a Bayesian network approach for identifying cis-regulatory modules likely to regulate tissue-specific expression. The network integrates predicted transcription factor binding site information, transcription factor expression data, and target gene expression data. At its core is a regression tree modeling the effect of combinations of transcription factors bound to a module. A new unsupervised EM-like algorithm is developed to learn the parameters of the network, including the regression tree structure.
Conclusion
Our approach is shown to accurately identify known human liver and erythroid-specific modules. When applied to the prediction of tissue-specific modules in 10 different tissues, the network predicts a number of important transcription factor combinations whose concerted binding is associated to specific expression.
Background
A cis-regulatory module (CRMs) is a DNA region of a few hundred base pairs consisting of a cluster of transcription factor (TF) binding sites [1]. By binding CRMs, transcription factors either enhance or repress the transcription of one or more nearby genes. Coordinated binding of several transcription factors to the same CRM is often required for transcriptional activation, thus allowing a very specific regulatory control.
High-throughput experimental identification of CRMs remains inaccessible, especially for distal enhancers. Methods like genomic localization assays (also known as ChIP-chip) using whole genome tilling arrays may soon improve the situation, but the cost of such extremely large arrays will limit their utilization. Because of this, several computational approaches have been developed for predicting cis-regulatory modules. Some attempt to identify regulatory modules with a particular function (e.g. muscle [2] or liver [3] specific CRMs, and many others [4–6]) by building or learning a model of the binding site content of such modules, based on a set of known modules. These methods generally obtain a reasonable specificity, but their applicability is limited to the few tissues, cell types, or conditions for which sufficiently many experimentally verified modules can be used for training. Others seek more generic signatures of cis-regulatory regions, like inter-species sequence conservation [7], sequence composition [8], or homotypic and heterotypic binding site clustering [9, 10]. These methods are more widely applicable, but their predictions may be of lesser accuracy, because they do not rely on any prior knowledge. Furthermore, the predictions made by these algorithms are not accompanied by any annotation regarding the putative function of the modules. The PReMod database [11] contains more than 100,000 human CRM computational predictions, mostly consisting of putative distal enhancers.
By adjoining other types of information to the predicted module information, additional insights can be gained into the function of specific modules. For example, in yeast, Beer and Tavazoie have used gene expression data to train a algorithm to predict expression data based on sequence information. In human, Blanchette et al. [12] and Pennacchio et al. [13] have used tissue-specific gene expression data from the GNF Altas2 [14] to identify certain transcription factors involved in tissue-specific regulation and Pennachhio et al. [13] have further developed models to predict the tissues-specificity of regulatory modules based on their binding site content. In this paper, we propose a new approach to the detection of tissue-specific cis-regulatory modules. Our algorithm uses a Bayesian network to combine binding site predictions and tissue-specific expression data for both transcription factors and target genes. It identifies the transcription factors and combinations thereof whose presence bound to a module appears to be resulting in tissue-specific expression. Our approach takes advantage of the facts that tissue-specific CRMs are likely 1) to be located next to genes expressed in that same tissue, 2) to contain many binding sites for TFs that are also expressed in that tissue, and (3) to contain binding sites whose presence in other modules also appears to be associated to tissue-specific expression. Our approach falls under the category of unsupervised learning, as it does not rely on any labeled training set or any type of prior knowledge regarding the TFs that may be important for a given tissue.
Importantly, the Bayesian network contains at its core a regression tree to represent the dependence between the regulatory activity of a CRM and the set of TFs predicted to bind it. A new unsupervised Expectation-Maximization-like algorithm is developed to infer the parameters of the network, including the structure of the regression tree. Our approach is related to that of Segal et al. [15, 16] but differs in that it takes advantage of available TF position weight matrices and TF expression data to allow tissue-specificity predictions. Moreover, based on the candidate modules predicted by PReMod, our approach is allowed to detect distal enhancers that are involved in tissue-specific expression.
We show that our method is able to accurately discriminate between known liver and erythroid-specific modules, even in the presence of a large fraction of modules with neither function, by discovering important combinations of transcription factors associated to these tissues. When applied to a larger set of putative modules and tissues, several known tissue-specific TFs were recovered, and many interesting new TF combinations were predicted to be linked to tissue-specific expression.
Methods
The goal of the method developed in this paper is to predict whether a given putative cis-regulatory module is responsible (at least in part) for the expression of a given gene in a particular tissue. Since the problem of predicting regulatory modules has already been studied extensively, we assume that a set of candidate CRMs has been identified in the genome under consideration and we focus on determining their tissue-specificity. We emphasize that many of these predicted CRMs are likely to be false-positives (i.e. they have no regulatory function whatsoever), and most are probably not specific to any tissue; our goal is to identify those that are. Given a putative CRM Mm, a gene G, and a tissue (or cell type) T, we want to determine whether module Mmup-regulates gene G in tissue T. (We focus only on the identification of enhancers, rather than repressors, because it is difficult to distinguish between repressed genes and genes that are not expressed due to the lack of activators.) To this end, we define a Bayesian network that is used to combine various types of evidence, including the putative transcription factor binding sites contained in Mm, the expression levels of the set of transcription factors predicted to bind Mm, and the expression level of gene G.
Importantly, and perhaps counter-intuitively, we train a single Bayesian network that will be applicable to predicting tissue-specific regulatory modules in all the tissues considered. This stems from the hypothesis that the enhancer activity of a module should depend only on its binding site content and on the expression levels of the transcription factors binding it, and not directly on the tissue considered. By allowing sharing regulatory mechanisms across tissues, we hope to improve our sensitivity to subtle regulatory mechanisms. One obvious drawback of this method is that unobserved entities like the presence or absence of tissue-specific transcriptional co-activators may affect the regulatory effect of a given module in different tissues even if the set of TFs bound to it does not change.
Typically, a Bayesian network consists of a set of observed variables, a set of unobserved variables, and an acyclic directed graph describing the direct dependencies between these. In this section, we first introduce the set of variables present in our network, which is depicted in Figure 1. We then describe the dependencies between these variables and the algorithms used to learn the parameters of the network.

The bayesian network used for predicting tissue-specific regulatory modules. See section 'Bayesian network variables' for a description of the variables, and section 'Bayesian network architecture' for a description of their dependencies.
Bayesian network variables
Let Φ = {Φ1,...,Φ|Φ|} be a set transcription factors, let be a set of tissue (or cell) types, let be the set of all known human protein-coding genes, and let be a set of predicted cis-regulatory modules. Since the notation describing the network requires many types of subscripts, we adopt the following convention: Right-subscripts refer to transcription factor indices; Right-superscripts refer to module indices; Left-superscripts refer to tissue indices; Left-subscripts refer to gene indices (for example, ). We start by defining the observed variables for our network, shown in unshaded ovals in Figure 1. More detailed definitions pertaining to the specific data set analyzed in this paper will be found in Section 'Data sets'. Consider the following domains of index variables: 1 ≤ m ≤ ||, 1 ≤ f ≤ |Φ|, 1 ≤ g ≤ ||, and 1 ≤ t ≤ ||.
-
is the real-number predicted affinity of transcription factor Φ f for module Mm. It should reflect our confidence that, provided factor Φ f is expressed, it will bind module Mm. It is a function of the number and the quality of Φ f 's predicted binding sites in Mm.
-
tF f is a boolean variable describing whether transcription factor Φ f is expressed in tissue tT.
-
is a boolean variable describing whether gene g is expressed in tissue tT.
To model the relationships between the observed variables, it is necessary to introduce a set of hidden variables.
-
is the actual state (active or inactive) of transcription factor Φ f in tissue tT. State may not equal the observed expression level tF f because of post-transcriptional regulation (e.g. activation due to external stimuli for nuclear receptors) or errors in the measurements of mRNA abundance.
-
is the actual transcriptional status (transcribed or not transcribed) of gene g G in tissue tT, which could be different from the observed mRNA abundance because of mRNA degradation or errors in the measurements of mRNA abundance.
-
is a boolean variable indicating whether, in tissue tT, module Mmis bound by sufficiently many copies of factor Φ f for this factor to achieve its function.
-
The fact that a module is bound by a transcription factor does not necessarily translate into this module being regulatorily active. Indeed, the presence of other transcription factors may be required for the module to become active. We represent the regulatory activity of module Mmin tissue tT by a boolean variable tRm, which takes the value 1 when the module Mmactively (and positively) regulates its gene. This is the variable whose value is of the most interest for predicting tissue-specific regulatory modules.
We acknowledge that using binary variables to represent expression levels and regulatory activity is a very crude approximation. Although all these variable should in theory be continuous, the quantitative relations between transcription factor expression levels, their binding affinity to a module, and the contribution of that module to the expression of the target gene remain poorly understood, so a more qualitative approach is preferable. Furthermore, due to the computational complexity of network inference, such a simplification was necessary. In fact, by reducing the size of the parameter search space, this simplification might actually be improving generalization from small data sets.
Bayesian network architecture
In a Bayesian network, dependencies between variables are modeled as directed edges connecting the cause to the effect. The conditional probability of a node given the value of its parent(s) is described by a set of parameters that are either fixed or learned from the data. When the variables at hand have a finite domain, these conditional probabilities can be represented by a conditional probability table (CPT).
Conditional distributions of E and F
The observed expression levels E and F depend on the true expression levels and respectively. Since all variables are boolean, the conditional probability tables are the following:

Here, α E and β E are the probabilities of false-positive and false-negative in the discretized gene expression data, respectively. We assume that these parameters are shared among all genes, i.e. expression measurement errors are equally likely for all genes. Similarly, α F and β F are the probabilities that the discretized expression measurement for a given factor does not reflect their actual regulatory potency. Again, these parameters are shared among all transcription factors, although this might to be inaccurate for factors like nuclear receptors, which require external signals for activation.
Conditional distribution of B
The probability of , the random variable that describes whether module Mmis bound by factor Φ f in tissue tT, depends on whether the factor is expressed in that tissue, and on the affinity of the factor for that module. We assume that the parameters describing this conditional probability are the same for all m and t, so we drop some subscripts and superscripts to write Pr[B f |A f , F f ]. We model this conditional probability indirectly, by instead modeling Pr[A f |B f = 1], the distribution of binding site affinities for a module that is bound, using a normal distribution with parameters μ f and that will be estimated during training. Since the mathematical derivation is tedious (but relatively simple), it is left in Appendix 1.
Conditional distribution of R using regression trees
The most challenging set of conditional probabilities to represent is that of tRm, which depends on the values of . Again, we assume the parameters that describe this dependency are the same for all tissues tT and all module Mm, so we drop these indices. This assumption is equivalent to saying that the regulatory effect of the binding of a certain set of transcription factors does not depend on the module bound, the gene being regulated, or the tissue type.
How should we represent the probability that a module is regulatorily active, given the set of transcription factors bound to it, i.e. Pr[R|B1,...,]? Given that all variables are boolean, this conditional probability can be represented by a × 2 CPT containing parameters. In our application where contains several hundred transcription factors, this is obviously not practical, because (1) the CPT would be too large to store, and (2) we would need a huge amount of training data to learn the parameters. We thus use a more compact representation for this CPT, based on regression trees [17]. A regression tree is a rooted tree whose internal nodes are labeled with tests on the value of some variable B f . See Figure 2 for a small example. For boolean variables (our case here), each node N tests whether the some variable takes the value true or false. Each leaf l of the tree is associated with a probability distribution Pr[R|l]. Let be the set of variable assignments obtained by following the path from the root to l. Let l(b1,...,) be the leaf reached when B1 = b1,..., = . Then, the regression tree defines a complete conditional probability distribution: Pr[R|B1 = b1,... = ] = p(l(b1,...,)). When many of the B i 's are irrelevant to R, the representation is much more compact than the standard CPT and can be estimated from less data. We will jointly refer to the tree topology, the node labelings, and the probability distributions at the leaves as the meta-parameter Ψ. Inferring Ψ will be the most significant difficulty of this approach.

Example of a regression tree representing a small 2-variable conditional probability table.
Conditional distribution of
The last set of dependencies is that of a gene's transcriptional activity on the regulatory activity of the neighboring regulatory modules. This raises the difficult question of determining which gene is being regulated by each module. This is relatively straight-forward when the module is located in the promoter region of a gene, but much less so when it is located 100 kb away from any gene. Here, for lack of more accurate information, we assume that a module Mmonly has the potential of regulating the gene g G whose transcription start site is the closest to it, denoted closest(Mm). Then the expression level of gene g G depends on regulators( g G) = {m|closest(Mm) = g G} = {r1, r2,...}. We will assume that the expression level of g G only depends on the number of its modules that are active, through a sigmoid function:
, where a and b are user-defined parameters (see Appendix 3).
Learning the network's parameters
Our Bayesian network contains a number of parameters whose values are not known a priori. We collectively refer to these parameters as . The network will be trained using the set of all pairs (module, tissue). Let A, E, and F be the set of all TF affinity data, all gene expression data, and all TF expression data, respectively, over all tissues considered. A typical approach to estimating the network's parameters is to seek the value Θ* that maximizes the joint likelihood of the observed variables, i.e. Θ* = argmaxΘPr[A, E, F].
An Expectation-Maximization algorithm can be used to learn the parameters Θ of the Bayesian network [18], whereby a local maximum of the likelihood function is reached by alternatively estimating the expected value of hidden variables given the observed variables and the current estimate Θ0, and then reestimating the maximum likelihood values for the parameters Θ. However, since Θ contains the tree structure, we cannot apply the standard EM algorithm for learning Bayesian networks, as this algorithm relies on the ability to analytically derive a maximum likelihood estimate for the parameters (see however [18]). Instead, a new EM-like algorithm with regression tree learning is developed to infer the tree within the network.
Estimating posterior probabilities for hidden variables
Our first step is to calculate the expectation (or equivalently, the probability of taking the value 1, since all hidden variables are binary), for all hidden variables, given the value of the observed variables. These posterior probabilities can be calculated using the formulas given in Appendix 2. The derivation of most of these formulas is fairly straight-forward, except for the calculations involving the regression tree. Computing can be done efficiently thanks to the regression tree representation.
Maximum likelihood parameter estimation
Once the posterior probabilities of the hidden variables are computed, maximum likelihood estimators for the parameters of the network can be derived as given in Appendix 3. Again, the regression tree representing the dependence of R on B1,..., poses significant challenge, as no efficient algorithm exists to choose the tree topology . Instead, we developed a new tree learning algorithm, which adapts ideas from standard decision tree algorithms (e.g. C 4.5 [19], J48 [20]). The problem at hand is novel and challenging for several reasons:
-
1.
Soft attributes: The input variables are binary variables, but their values remain unknown at any given iteration of the EM-like algorithm. Only their distribution Pr[|A, E, F] is known for each m, f and t, given the current estimate of the parameters Θ.
-
2.
Soft labels: The values of the target variables tRmare also unknown, but their distribution Pr[tRm|A, E, F] is known.
Learning regression trees from probabilistic instances
Most decision tree learning algorithms are based on a greedy tree-growing approach trying to find the tree that minimizes the number of misclassifications [21]. Our tree learning algorithm is an adaptation of the standard approach using information gain as a method to select which attribute to select to split a node. Consider a node N that is currently a leaf and that we are considering splitting based on some attribute B i . The weight of a probabilistic instance is the probability of the path from the root to N, under the attribute probability distributions given by x.
More precisely,
We can now define the weighted entropy at node N as:
where , and totalWeight(N) = ∑ t ∑ m weight N (m, t). Then, the information gain obtained by splitting a leaf N with attribute B i to obtain two new leaves N' and N" is defined as
The attribute B i with the largest weighted information gain is chosen as label for N and corresponding children nodes N' and N" are added. The tree grows this way until no pair of node and attribute yields a positive information gain. This is a very loose stopping criterion and trees learned this way tend to be very large.
In order to avoid the problem of overfitting, a method called reduced-error pruning is used [21]. It uses a separate validation data set to prune the tree, and each split node in the tree is considered to be a candidate for pruning. When pruning a node, a operation called subtree replacement is performed, which involves removing the subtree rooted at that node and replacing the subtree with a single leaf. Whether pruning is performed depends on the classification accuracy obtained by the unpruned tree and by the pruned tree over the validation set. Pruning will cause the accuracy over the training data set to decrease; but it may increase the accuracy over the test data set.
Results
Our approach was used to identify tissue-specific CRMs in human. First, we show, using a small set of experimentally verified tissue-specific CRMs, that our approach is able to discriminate between modules involved in different tissues. Then, we apply our method to a larger data set consisting of more than 6000 putative CRMs associated to genes specifically expressed in one of ten tissues, and show that interesting combinations of transcription factors can be linked to tissue-specific expression.
Data sets
We used a set of cis-regulatory modules predicted in the human genome by Blanchette et al. [12], based on a set of 481 position weight matrices from Transfac 7.2 [22]. The modules are available from the PReMod database [11]. Criteria used for the PReMod predictions include inter-species conservation of binding site predictions and homotypic clustering of binding sites. The complete data set consists of more than 100,000 predicted CRMs, but only subsets of those were used (see below). For each predicted module Mm, the predicted binding affinity is represented by the negative logarithm of the p-value of the binding site weighted density for factor Φ f in module Mm, as reported in PReMod. Gene expression data came from the GNF Atlas 2 data set [14], downloaded from the UCSC Genome Browser [23]. A gene g G was identified as "expressed" (i.e. = 1) if and only if its expression level was at least two standard deviations above its mean expression level, over the 79 tissues for which data was available.
Only 231 of the 481 Transfac PWMs were confidently linked to transcription factors for which GNF expression data is available. Only these || = 231 PWMs were considered in our analysis. Some transcription factors are actually linked to several different PWMs, but our approach actually seems to take advantage of this to improve the quality of the predictions (see below).
Validation experiments
We first use a set of experimentally verified tissue-specific CRMs, together with a set of negative control regions, to validate our algorithm. To further evaluate the performance of our approach, we compare our results with the results obtained with several simpler classifiers.
Validation data sets
To demonstrated the ability of our approach to identify tissue-specific regulatory modules, we used it to discriminate between known liver-specific CRMs, known erythroid-specific CRMs, and other modules not likely to be involved in these two cell types. Each validation data set was composed of five subsets:
-
1.
knownLiver: 11 experimentally verified liver-specific modules [3].
-
2.
knownErythroid: 22 experimentally verified erythroid-specific modules [24].
-
3.
putativeLiver: A set of 31 PReMod modules located in the vicinity of the genes associated to the knownLiver modules. These modules are possibly involved in liver-specific regulation and are included only to help the Bayesian network learning the association between a module's binding site composition and tissue-specificity of the target gene.
-
4.
putativeErythroid: A set of 46 PReMod modules similar to (3) but for erythroid.
-
5.
negative: For each knownErythroid or knownLiver module with associated closest gene g, a set of r neg (see below) PReMod modules associated to genes that are expressed in neither erythroid nor liver is randomly selected and artificially associated to gene g. These are modules that, if placed in the vicinity of gene g, would be unlikely to cause liver or erythroid-specific expression.
The ratio r neg of the number of negative modules to the number of known modules determines in part the difficulty of the classification task. Two types of validation data sets were thus created: In our 1X experiment (see below), we used r neg = 1, whereas in our 2X data set, we used r neg = 2.
Each 1X data set thus contains 143 modules, each of which was considered as a possible liver or erythroid specific. The complete data set consists of 2 × 143 = 286 module-tissue pair, of which 11 + 22 = 33 are positive examples, 99 are negative examples (all the knownLiver modules when considered in the erythroid cell type, all the knownErythroid modules when considered in liver, and all the negative modules in both tissues). The 2X datasets are similar, except that they are noisier because they contain 165 negative examples.
Three simple classifiers
To assess the quality of our method, we compare it to three other simpler approaches. The first classifier, called the expressionOnly classifier, simply predicts that any module located next to a gene that is expressed in a given tissue is a tissue-specific module for that tissue. That is, the binding site content of the module is ignored, and only the expression g E is used to make the prediction.
The second simple classifier, called SupervisedNaiveBayes, is a classical supervised Naive Bayes approach that takes as input a simplified, observable version of the B variables, where we set = F m ·Af, as well as the expression of the target gene and is trained to distinguish between labeled positive and negative examples (see Appendix 4 for the complete details). Finally, the third simple classifier, called NaiveBayesInNet, is a version of our Bayesian Network classifier in which the regression tree representing the conditional probability of R is replaced by a Naive Bayes classifier, but where the rest of the structure is preserved. See Appendix 5 for more details.
Validation results
One hundred different runs of our EM-like algorithm were done on 1X and 2X datasets, each time with a different sample of negative modules. Each run used 100 EM-like iterations (taking approximately 10 minutes of running time), which was sufficient to achieve convergence, although different runs converge to slightly different likelihoods and regression trees (see Additional File 1). Since we do not know which of the putativeLiver and putativeErythroid CRMs are actually tissue-specific modules, we evaluate the performance of our algorithm based only on the positive and the negative modules. For each run, the network with the best likelihood over 100 EM-like iterations is used to compute Pr[tRm|A, E, F] for all examples and a module-tissue pair is predicted positive if this probability exceed some threshold t. The resulting precision-recall curve, averaged over all 100 runs, is shown in Figure 3, for both the 1X and 2X data set.

The precision v.s. recall curve for the 1X (left) and 2X (right) data sets, where precision = TP/(TP + FP) and recall = TP/(TP + FN). The blue curve (diamond markers) is generated from the results of our approach, the brown curve (× markers) is generated from the results of the Supervised-NaiveBayes approach (see Appendix 4), and the green curve (circle markers) is generated from the results of the NaiveBayesInNet classifier (see Appendix 5). The pink triangle shows the result obtained by the expressionOnly classifier. Error bars denote one standard deviation of the precision, over 100 random choices of negative examples. The increase in the standard deviation on precision at lower recall is due to the small number of predictions made for these thresholds.
Since 13 out of the 33 known CRMs have target genes expressed neither in liver nor in erythroid (based on our discretization of expression data), the ExpressionOnly classifier yields a recall = 60.6% and precision = 50% on the 1X data set, but only precision = 33% on the 2X data set.
As seen from the curves, our method significantly outperforms both the Naive Bayes-based approaches for mid- to high-precision predictions. Our method can improve the precision to 72% for the 1X data sets and 66.2% for the 2X data sets. Notice that the highest precision for the 2X data sets remains close to that for the 1X data sets, although almost twice as many negative examples are considered. This indicates that our approach provides a way to improve the precision of prediction by combining the sequence data and the expression data.
Regression trees
Figure 4 shows the regression trees generated from one run for the 1X and 2X data sets. Each internal node tests the value of an attribute B f , which indicates whether factor Φ f is predicted to bind the module in the tissue under consideration. Each leaf shows the conditional probability predicted, which is the probability of R = 1 on the condition specified by the path from to root to the leaf.

The regression tree generated by the iteration with the best likelihood for a 1X (top) and 2X (bottom) data sets. Internal nodes corresponding to liver-specific transcription factors are colored yellow, and those corresponding to erythroid-specific factors are red.
The tree structure indicates what are the most important TFs or combinations of TFs for explaining liver-specific and erythroid-specific expression. Our algorithm successfully detects most known liver-specific TFs and combinations of thereof, like HNF1 + HNF4, HNF1 + C/EBP, and HNF4 + C/EBP, which are reported in the literature [3]. The erythroid-specific TF GATA1 is also reported in the trees. The trees do not contain many erythroid-specific nodes, firstly because there are only two TFs (GATA1 and NF-E2) that are erythroid-specific based on our expression data, and secondly because NF-E2 has very few predicted binding sites on the genome. We observe from the trees that the leaves associated with TF combinations usually have higher regulatory probabilities than the leaves associated with individual TFs. This indicates that the ability to identify TF combinations is key to being able to identify cis-regulatory modules. We emphasize that the trees were obtained without any prior information about which of the 231 PWMs used are involved in liver or erythroid-specific expression.
Notice that TF PPAR is reported in our trees. PPAR is indeed an important factor regulating expression in liver [25], but was absent from Krivan and Wasserman's paper [3] from which we obtain the known liver-specific CRMs. Most importantly, the expression of PPAR is low in both liver and erythroid, so erythroidF PPAR = liverF PPAR = 0. This shows that our approach is robust to noise in the expression data of TFs, provided the association between the binding sites in modules and the target gene's expression is sufficiently high. Finally, we note the unexpected selection of several different matrices for the same transcription factor along the same path in the tree (for example C/EBP M770 and M190 on the tree obtained for the 1X data set on Figure 3). This is caused by the fact that these matrices are quite actually different from each other, but the presence of sites for both matrices increases the association to the target gene's expression.
Genome-wide CRM prediction in ten tissues
We next extended our analysis to ten different tissues from the GNF Atlas2: = {brain, erythroid, thyroid, pancreatic islets, heart, skeletal muscle, uterus, lung, kidney, and liver}. 923 genes are specifically expressed (i.e. = 1) in at least one of these tissues and a total of 6278 modules are associated to these genes. We thus trained our Bayesian network on a set of 10 × 6, 278 = 62, 780 (module, tissue) pairs. Ten parallel runs of 100 EM-like iterations were performed from different random initializations, each taking approximately 24 hours.
The regression tree obtained obtained from the best run is shown in Figure 5. We can clearly observe from the tree that the positive assignments along each path leading to a leaf typically consists of TFs expressed in the same tissue. Several known tissue-specific combinations of TFs are recovered in the tree, such as C/EBP + HNF1 and C/EBP + HNF4 in liver. Also, many new and potentially meaningful TF combinations are predicted, such as C/EBP + AR in liver and Tax/CREB + GATA1 in erythroid.

Regression tree obtained from the best of ten runs on the set of 6,278 modules and 10 tissues. Nodes are colored based on the tissue in which a particular factor is expressed.
The tree only contains the TFs expressed in four tissues: liver, erythroid, heart, and skeletal muscle. The other six tissues are not represented in the tree because of one of the following reasons: (1) The TFs that regulate the genes expressed in those tissues have low expression levels. (2) These TFs do not have strict requirements for sequence affinity, so the binding scores of their matrices are low. It is also possible that there are no PWMs for such TFs. (3) The expression of genes in those tissues are controlled by post-transcriptional regulation instead of tissue-specific TFs.
The complete set of tissue-specificity predictions are available at http://www.mcb.mcgill.ca/~xiaoyu/tissue-specificModule.
Statistical analysis of TF combinations
The regression trees obtained in the 10 runs vary substantially in their structure but share many of their factors and combination of factors. The frequency with which factors or combination of factors are found in these trees is an indication of their role in regulating tissue-specific expression. A pair of factors is said to co-occur in a regression tree if the tree contains a path along which both factors take value 1. As seen in Tables 1 and 2, several factors and pairs of factors are consistently identified as part of the tree. Most TFs found are either known to be directly involved in tissue-specific regulation (in bold in Table 1, or to be essential for the expression of certain genes in the given tissues, but to also have other non-tissues-specific roles (normal font in Table 1).
Predicting gene tissue-specificity
To further validate our module tissue-specificity predictions, we investigated whether a gene's tissue-specific fine-grain expression level could be predicted based on the modules regulating it. To this end, for each tissue t, we separated genes between highly expressed ) and low expressed ( = 0). Let be the maximum of the predicted regulatory activity tRmof the modules associated to gene g. We asked whether is predictive of the raw, non-thresholded expression level of gene g. In the case of genes with = 0, such a correlation would show that we are able to detect tissue-specific genes even if their expression level is below the threshold. For genes with = 1, this correlation would show that genes with very high tissue-specific expression levels are associated to stronger module predictions than those that barely meet our threshold. We note that in both cases, such a correlation could not be explained by any kind of training artifact, since raw expression data is not part of the input.
Considering genes showing tissue-specific expression ( = 1), we find that eight of the ten tissues considered (all but "whole brain" and "erythroid") exhibit a positive correlation between and the raw gene expression. Somewhat surprinsingly, the correlation is strongest for thyroid (p-value = 0.028) and skeletal muscle (p-value = 0.015), two factors that were relatively poorly represented in our regression tree. Among genes with = 0, the correlation is weaker but is positive in seven of the ten tissues (all except heart, skeletal muscle, and liver). These results indicate that our predictions yield a weak predictor of gene tissue-specificity. Clearly, it is easier to predict modules responsible for a gene's observed tissue-specificity than to predict the tissue-specificity of a gene based on its modules.
Discussion and conclusion
The approach we introduced here is the first to integrate binding site predictions and tissue-specificity of expression of both transcription factors and target genes to predict cis-regulatory modules involved in regulating tissue-specific gene expression. By introducing a regression tree at the heart of the network and deriving practical algorithms to train it, we are able to accurately identify important combinations of transcription factors regulating gene expression in a tissue-specific manner. The algorithms derived for learning this type of network will undoubtedly be applicable to a wide range of problems.
Many of the choices made in the design of the Bayesian network were made for computational practicality reasons. As we improve the learning algorithm, it will become possible to use real-numbered expression measurements.
Furthermore, our network could easily be extended by introducing additional sources of information as observed variables. For example, ChIP-chip and other binding assay data, when available, can be used to affect our belief in . Reporter assays and DNA accessibility assays could be used to modify our belief in tRm. If modeled correctly, these types of experimental data may greatly increase the accuracy of our predictions, not only for the modules or the factors for which data is available, but also for other regions or factors associated to similar functions.
The approach we described is potentially applicable to a wide range of data sets. While the relative inefficiency of the current learning algorithm prevented us from analyzing the complete set of tissue-specific expression from GNF, it is clear that this analysis, involving 79 tissues, would yield a wealth of information. Another possible application is to identify and characterize cis-regulatory modules involved in time and tissue-specific regulation during fish development. The large body of in situ hybridization data available in zebrafish [26] would provide an excellent basis for this analysis.
Appendix 1. Calculation of Pr[B f |A f , F f ]
Pr[B f |A f , F f ] is the probability of TF Φ f binding a genomic region, given its observed expression F f and its binding affinity A f for the region. Modeling this relationship is challenging because it is unclear how B f , a binary variable, should depend on A f , a continuous variable, in the presence of the observed expression F f . For this reason, we derive this probability from a set of other probabilities distributions that are easier to model, specifically Pr[A f |B f = 1], the affinity score distribution of sites that are bound.
Recall that is defined as the actual expression of factor Φ f . Note first that
for some appropriately chosen constants Z and Z'. The distribution of Pr[F f |] is described in Section 'Conditional distributions of E and F', and the prior probability Pr[] is approximated by the prior probability of the observed variable Pr[F f ]. So all that remains is to define Pr[A f |B f , ] and Pr[B f |].
Because a TF can bind only if it is expressed, we have

When = 0, the event B f = 0 yields no information on A f , so

where the prior probability Pr[A f ] is estimated from the data using a histogram approach.
Notice that

We thus obtain
where

and where

We assume that Pr[A f |B f = 1] follows a normal distribution with parameters μ f and that are optimized during the EM-like algorithm (see Appendix 3). Pr[F f |] and Pr[] have all been previously defined.
Finally, Pr[B f |] is represented by a fixed CPT:

where γ = 0.01 is a parameter that indicates the prior probability that an expressed TF will bind a generic genomic region.
Appendix 2. Formulas for E-step
Calculation of Pr[Rm|A, E, F]
Let X be the set of modules associated with the same gene g. Let S = ∑r∈XRr, we where
where
-
The regression tree allows an efficient computation of the first sum:
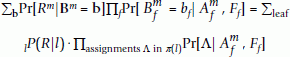
-
Pr[|, F f ] has been defined in Appendix 1;
-
Pr[ g E| g ] is represented by a CPT described in Section 'Conditional distributions of E and F';
-
Pr[ g |S = s] is defined by the sigmoid function 1/(1 + e-b(s-a)).
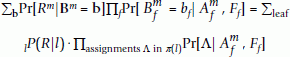
Further noting that
Pr[S = s|Rm, AX-m, F] = Pr[∑r∈X-mRr= s - Rm|AX-m, F], we can calculate Pr[S = s|Rm, AX-m, F] by using a simple dynamic programming.
Calculation of Pr[|A, B, F]
Note that Pr[|, F f ] has been defined in Appendix 1. Furthermore, we can estimate Pr[Rm|] from the data
where Pr[|A, E, F] takes the values calculated from the previous iteration.
We thus get
where Pr[|, F f ] is obtained as in Appendix 1 and
Calculation of Pr[|A, F, E] and Pr[|A, F, E]
Although is a hidden variable, its posterior probability distribution does not need to be estimated, because we sum over all its possible values when computing Pr[Rm|A, F, g E]. The same holds for in Pr[B f |A f , F f ].
Appendix 3. Parameter re-estimation (M-step)
Pr[A f |B f = 1] is assumed to follow a normal distribution N(μ f , ).
Parameters μ f and σ f are re-estimated as follows:
In order to avoid overstepping the local maximum, we use small steps when updating the values of μ f and σ f . Instead of replacing the old values with the new values calculated from Equation 1, we use a hybrid of the old values and the new values, weighted according to the step size.
where α = 0.1 is the step size.
The following parameters have values that remain fixed throughout the execution of the EM-like algorithm. Their value has been chosen empirically to optimize the quality of the results.
-
1.
Parameters for Pr[E|] and Pr[F|]: α E = β E = α F = β F = 0.1
-
2.
Parameters for :
a = 0.8, b = 10 in validation experiments (small data sets), and
a = 0.4, b = 10 in discovery experiments (large data set).
-
3.
Parameters for Pr[B f |]: γ = 0.01.
Appendix 4. The SupervisedNaiveBayes classifier
A naive Bayes classifier was trained to discriminate between positive and negative (module, tissue) pairs. First, the affinity is discretized as 1 if and only if its value is at least one standard deviation above the mean of A i , over all 100,000 putative modules from PReMod. The Naive Bayes network takes as input the following set of attributes: , and E g . The precision-recall curves from Figure 3 were the result of a 11-fold cross-validation experiment.
Appendix 5. The NaiveBayesInNet classifier
The NaiveBayesInNet classifier is a Bayesian Network identical to the main classifier presented in this paper, except that a Naive-Bayes-like approach replaces the probability tree representing Pr[R|B1,...,BΦ]. More specifically, it assumes Pr[R|B1,...,BΦ] = ∏f=1...ΦPr[B f |R]/Z.
At each iteration of the EM-like algorithm, Pr[B f |R] is estimated as . Then, estimating Pr[R|A, F, E] requires a summation over all possible values of the B variables (the simplification afforded by the regression tree cannot be applied here). To make the computation practical, we instead fix the value of the B to their maximum likelihood estimates and use these fixed values to estimate Pr[R|A, F, E]. The approach was trained and evaluated using exactly the same methodology as for the Bayes network approach using regression trees.
References
Davidson EH: Genomic regulatory systems: development and evolution. Academic Press; 2001.
Wasserman W, Fickett J: Identification of regulatory regions which confer muscle-specific gene expression. J Mol Biol 1998, 278: 167–81. 10.1006/jmbi.1998.1700
Krivan W, Wasserman W: A predictive model for regulatory sequences directing liver-specific transcription. Genome Res 2001,11(9):1559–66. 10.1101/gr.180601
Aerts S, Loo PV, Thijs G, Moreau Y, Moor BD: Computational detection of cis-regulatory modules. Bioinformatics 2003,19(Suppl 2):II5-II14.
Bailey TL, Noble WS: Searching for statistically significant regulatory modules. Bioinformatics 2003,19(Suppl 2):II16-II25.
Sinha S, van Nimwegen E, Siggia ED: A probabilistic method to detect regulatory modules. Bioinformatics 2003,19(Suppl 1):i292–301. 10.1093/bioinformatics/btg1040
Prabhakar S, Poulin F, Shoukry M, Afzal V, Rubin E, Couronne O, Pennacchio L: Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res 2006,16(7):855–863. 10.1101/gr.4717506
Taylor J, Tyekucheva S, King D, Hardison R, Miller W, Chiaromonte F: ESPERR: Learning strong and weak signals in genomic sequence alignments to identify functional elements. Genome Res 2006,16(12):1596–1604. 10.1101/gr.4537706
Philippakis AA, He FS, Bulyk ML: Modulefinder: a tool for computational discovery of cis regulatory modules. Pac Symp Biocomput 2005, 519–30.
Johansson O, Alkema W, Wasserman W, Lagergren J: Identification of functional clusters of transcription factor binding motifs in genome sequences: the MSCAN algorithm. Bioinformatics 2003,19(Suppl 1):i169–76. 10.1093/bioinformatics/btg1021
Ferretti V, Poitras C, Bergeron D, Coulombe B, Robert F, Blanchette M: PReMod : a database of genome-wide mammalian cis-regulatory module predictions. Nucleic Acids Res 2007, (35 Database):D122–6. 10.1093/nar/gkl879
Blanchette M, Bataille AR, Chen X, Poitras C, Lananiere J, Lefebvre C, Deblois G, Giguere V, Ferretti V, Bergeron D, Coulombe B, Robert F: Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Research 2006,16(5):656–668. 10.1101/gr.4866006
Pennacchio L, Loots G, Nobrega M, Ovcharenko I: Predicting tissue-specific enhancers in the human genome. Genome Res 2007,17(2):201–211. 10.1101/gr.5972507
Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB: A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 2004,101(16):6062–7. 10.1073/pnas.0400782101
Segal E, Yelensky R, Koller D: Genome-wide discovery of transcriptional modules from DNA sequence and gene expression. Bioinformatics 2003,19(Suppl 1):i273–82. 10.1093/bioinformatics/btg1038
Segal E, Barash Y, Simon I, N F, Koller D: From Promoter Sequence to Expression: A Probabilistic Framework. Proc 6th Inter Conf on Research in Computational Molecular Biology (RECOMB) 2002.
Boutilier C, Friedman N, Goldszmidt M, Koller D: Context-specific independence in Bayesian networks. Proc Twelfth Conf on Uncertainty in Artificial Intelligence (UAI-96) 1996.
Dempster A, Laird N, Rubin D: Maximum likelihood from incomplete data via the EM algorithm. J of the Royal Statistical Society, Series B 1977, 39: 1–38.
Quinlan J: C4.5: Programs for machine learning. Morgan Kaufmann; 1993.
Witten I, Frank E: Data Mining: practical machine learning tools with Java implementations. Morgan Kaufmann; 2000.
Mitchell TM: Machine learning. McGraw-Hill; 1997.
Matys V, Fricke E, Geffers R, Gössling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel A, Kel-Margoulis O, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Münch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E: TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 2003, 31: 374–8. 10.1093/nar/gkg108
Karolchik D, Baertsch R, Diekhans M, Furey T, Hinrichs A, Lu Y, Roskin K, Schwartz M, Sugnet C, Thomas D, Weber R, Haussler D, Kent W, Kent W: The UCSC Genome Browser Database. Nucleic Acids Res 2003, 31: 51–4. 10.1093/nar/gkg129
Podkolodnaya OA, Stepanenko IL: The ESRG-TRRD: database of genes with specific transcription regulation in erythroid cells.1998. [http://wwwmgs.bionet.nsc.ru/mgs/papers/podkolodnaya/esg-trrd]
Yoshikawa T, Ide T, Shimano H, Yahagi N, Amemiya-Kudo M, Matsuzaka T, Yatoh S, Kitamine T, Okazaki H, Tamura Y, Sekiya M, Takahashi A, Hasty AH, Sato R, Sone H, Osuga JI, Ishibashi S, Yamada N: Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. I. PPARs suppress sterol regulatory element binding protein-1c promoter through inhibition of LXR signaling. Mol Endocrinol 2003,17(7):1240–54. 10.1210/me.2002-0190
Sprague J, Bayraktaroglu L, Clements D, Conlin T, Fashena D, Frazer K, Haendel M, Howe D, Mani P, Ramachandran S, Schaper K, Segerdell E, Song P, Sprunger B, Taylor S, Slyke CV, Westerfield M: The Zebrafish Information Network: the zebrafish model organism database. Nucleic Acids Res 2006, (34 Database):D581–5. 10.1093/nar/gkj086
Krivan W, Wasserman W: A predictive model for regulatory sequences directing liver-specific transcription. Genome Research 2001,11(9):1559–1566. 10.1101/gr.180601
Eagon P, Elm M, Stafford E, Porter L: Androgen receptor in human liver: characterization and quantitation in normal and diseased liver. Hepatology 1994,19(1):92–100.
Lecointe O, Bernard K, Naert V, Joulin C, Larsen P, Romej , D MM: GATA-and SP1-binding sites are required for the full activity of the tissue-specific promoter of the tal-1 gene. Oncogene 1994, 9: 2623–2632.
Humbert P, Rogers C, Ganiatsas S, Landsberg R, Trimarchi J, Dandapani S, Brugnara C, Erdman S, Schrenzel M, Bronson R, Lees J: E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell 2000,6(2):281–91. 10.1016/S1097-2765(00)00029-0
Bockamp E, McLaughlin F, Gottgens B, Murrell A, Elefanty A, Green A: Distinct Mechanisms Direct SCL/tal-1 Expression in Erythroid Cells and CD34 Positive Primitive Myeloid Cells. Journal of Biological Chemistry 1997,272(13):8781–8790. 10.1074/jbc.272.13.8781
Blobel G, Nakajima T, Eckner R, Montminy M, Orkin S: CREB-binding protein cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc Natl Acad Sci USA 1998,95(5):2061–2066. 10.1073/pnas.95.5.2061
Welch J, Watts J, Vakoc C, Yao Y, Wang H, Hardison R, Blobel G, Chodosh L, Weiss M: Global regulation of erythroid gene expression by transcription factor GATA-1. Blood 2004,104(10):3136–3147. 10.1182/blood-2004-04-1603
Dufour C, Wilson B, Huss J, Kelly D, Alaynick W, Downes M, Evans R, Blanchette M, Giguere V: Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metabolism 2007,5(5):345–56. 10.1016/j.cmet.2007.03.007
Zhu W, TomHon C, Mason M, Campbell T, Shelden E, Richards N, Goodman M, Gumucio D: Analysis of Linked Human epsilon and gamma Transgenes: Effect of Locus Control Region Hypersensitive Sites 2 and 3 or a Distal YY1 Mutation on Stage-Specific Expression Patterns. Blood 1999,93(10):3540–9.
Crestani M, De Fabiani E, Caruso D, Mitro N, Gilardi F, Vigil Chacon A, Patelli R, Godio C, Galli G: LXR (liver X receptor) and HNF-4 (hepatocyte nuclear factor-4): key regulators in reverse cholesterol transport. Biochem Soc Trans 2004,32(Pt 1):92–6. 10.1042/BST0320092
Peterkin T, Gibson A, Loose M, Patient R: The roles of GATA-4, -5 and -6 in vertebrate heart development. Semin Cell Dev Biol 2005,16(1):83–94. 10.1016/j.semcdb.2004.10.003
Reimold A, Etkin A, Clauss I, Perkins A, Friend D, Zhang J, Horton H, Scott A, Orkin A, Byrne M, Grusby M, Glimcher L: An essential role in liver development for transcription factor XBP-1. Genes Dev 2000,14(2):152–157.
Charron J, Malynn B, Fisher P, Stewart V, Jeannotte L, Goff S, Robertson E, Alt F: Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev 1992, 6: 2248–2257. 10.1101/gad.6.12a.2248
Acknowledgements
We wish to thank Doina Precup, Eric Blais, Emmanuel Mongin, Francois Pepin, and two anonymous reviewers for their useful comments. XC was funded by Genome Quebec Comparative and Integrative Genomics.
This article has been published as part of BMC Bioinformatics Volume 8 Supplement 10, 2007: Neural Information Processing Systems (NIPS) workshop on New Problems and Methods in Computational Biology. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/8?issue=S10.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XC designed and implemented the prediction algorithms, obtained all the results presented, and participated to the manuscript redaction. MB contributed to the original idea, the mathematical formulation and the redaction. All authors read and approved the final manuscript.
Electronic supplementary material
12859_2007_1979_MOESM1_ESM.jpg
{kind=link}
Additional file 1: The logarithms of the likelihoods for the 2X validation experiments in three different randomly selected runs. Different colors represent different runs. (JPG )
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chen, X., Blanchette, M. Prediction of tissue-specific cis-regulatory modules using Bayesian networks and regression trees. BMC Bioinformatics 8 (Suppl 10), S2 (2007). https://doi.org/10.1186/1471-2105-8-S10-S2
Published:
DOI: https://doi.org/10.1186/1471-2105-8-S10-S2