- Methodology Article
- Open access
- Published:
CellECT: cell evolution capturing tool
BMC Bioinformatics volume 17, Article number: 88 (2016)
Abstract
Background
Robust methods for the segmentation and analysis of cells in 3D time sequences (3D+t) are critical for quantitative cell biology. While many automated methods for segmentation perform very well, few generalize reliably to diverse datasets. Such automated methods could significantly benefit from at least minimal user guidance. Identification and correction of segmentation errors in time-series data is of prime importance for proper validation of the subsequent analysis. The primary contribution of this work is a novel method for interactive segmentation and analysis of microscopy data, which learns from and guides user interactions to improve overall segmentation.
Results
We introduce an interactive cell analysis application, called CellECT, for 3D+t microscopy datasets. The core segmentation tool is watershed-based and allows the user to add, remove or modify existing segments by means of manipulating guidance markers. A confidence metric learns from the user interaction and highlights regions of uncertainty in the segmentation for the user’s attention. User corrected segmentations are then propagated to neighboring time points. The analysis tool computes local and global statistics for various cell measurements over the time sequence. Detailed results on two large datasets containing membrane and nuclei data are presented: a 3D+t confocal microscopy dataset of the ascidian Phallusia mammillata consisting of 18 time points, and a 3D+t single plane illumination microscopy (SPIM) dataset consisting of 192 time points. Additionally, CellECT was used to segment a large population of jigsaw-puzzle shaped epidermal cells from Arabidopsis thaliana leaves. The cell coordinates obtained using CellECT are compared to those of manually segmented cells.
Conclusions
CellECT provides tools for convenient segmentation and analysis of 3D+t membrane datasets by incorporating human interaction into automated algorithms. Users can modify segmentation results through the help of guidance markers, and an adaptive confidence metric highlights problematic regions. Segmentations can be propagated to multiple time points, and once a segmentation is available for a time sequence cells can be analyzed to observe trends. The segmentation and analysis tools presented here generalize well to membrane or cell wall volumetric time series datasets.
Background
Fluorescent microscopy datasets composed of nuclear and membrane (or cell wall) channels pose problems to automated image analysis algorithms, as seen in the examples in Fig. 1. To address these challenges we propose an interactive segmentation and analysis tool that guides the user to quickly correct potential errors and adaptively propagate these corrections through the entire dataset, thus providing a robust framework for further quantitative image analysis and results validation.

Image segmentation challenges. a Reconstructed cross-section of a SPIM light sheet microscopy volume of the ascidian P. mammillata showing an artifact in which as many as five nuclei appear connected. This makes it difficult for existing nuclei detection methods to properly segment. b Weak signal in the membrane channel in lower z slices of a confocal microscopy image. c Inconsistent signal strength in the cell wall channel of a z slice through a confocal microscopy image of Arabidopsis thaliana (image courtesy Elliot Meyerowitz Lab, Division of Biology, California Institute of Technology). d Cells with interrupted membrane which share cytoplasm, as in this example of the Caenorhabditis elegans gonad cells [32]. Watershed segmentation methods will have difficulty segmenting such structures due to leakage. e Sperm cells appear in the nuclei channel resulting in false positives for a nuclei detector [32]. f Dividing P. mammillata cell SPIM images that show up as large nuclei
Interactive segmentation has gained significant interest in the bio-imaging community in recent years. For example, [1] proposes an interactive learning approach for segmentation of histological images. Ilastik is a widely used interactive segmentation and classification tool [2]. Other tools are specifically targeted to, for example electron microscopy images [3] or for segmentation of clusters of cells such as [4] which classifies pixels based on the geodesic commute distance and spectral graph theory. The user-guided segmentation algorithm in [5] is aimed at 3D nuclei segmentation and integrates multiple nuclei models simultaneously. The software introduced in [6] offers interactive visualization and analysis tools which enable users to create a processing pipeline for microscopy applications, from image filtering to segmentation and analysis. The work of [7] uses an active contour approach based on parametrized B-splines for interactive 3D segmentation. A conditional random field whose underlying graph is a watershed merging tree is trained in the interactive segmentation approach of [8] and is applied to segmentation of neuronal structures in electron microscopy data.
Here we introduce an interactive cell analysis application called CellECT (Fig. 2), which consists of a segmentation component and an analysis component. The user can modify a label map that is obtained using seeded Watershed [9], by adding, removing or modifying segments. The algorithm aims at obtaining correct segmentation with minimum user interaction. We define an adaptive metric we call cellness which is trained to highlight the regions where the segmentation is likely to be incorrect and may require the user’s attention. Additionally, the algorithm can offer specific suggestions. Segmentation results can then be propagated to other time points in the 3D+t dataset. Furthermore, CellECT provides an analysis component which summarizes the changes in various cell measurements over the time sequence. A user-friendly interface allows for easy workspace management, including the import of 3D or 3D+t TIFF stacks with any additional information (e.g. metadata such as scale, nuclei detection, or anterior-posterior axis of the specimen), opening an existing workspace for continuing work, or appending two existing workspaces to concatenate time points from separate TIFF files.

CellECT software screenshots. CellECT enables the interactive segmentation and analysis of 3D+t microscopy membrane (or cell wall) volumes. Screenshots of CellECT’s main interface (left-most), the interactive segmentation tool (left-middle), and analysis module (right) are shown above
The primary contributions include: (1) an interactive segmentation tool that manages user guidance markers in the geodesic image space, (2) an adaptive cellness metric that learns from user-feedback and computes/maintains a probabilistic belief about the quality of a cell’s segmentation and a method to make suggestions to the user, (3) the ability to propagate user corrections to other time points, and (4) an analysis component which facilitates quantitative observation about the organism’s development changes over a time sequence. These algorithms and features are packaged into an open source software application. We utilize this software for the analysis of a 3D+t confocal microscopy dataset of the ascidian P. mammillata consisting of 18 time point, a 3D+t SPIM dataset of P. mammillata consisting of 192 time points, and a dataset of eight 2D confocal microscopy slices of A. thaliana consisting of 112 pavement cells.
Methods
CellECT overview
CellECT is an application for interactive segmentation and analysis of 3D+t microscopy datasets containing cell boundary information (e.g., plasma membrane or cell wall). Its features include:
Workspace management: CellECT allows users to import a dataset in TIFF format along with other optional information such as image metadata, seed points (e.g. nuclei) obtained with an external tool (e.g. a nuclei detector application), or sample orientation (e.g., anterior-posterior axis). CellECT then creates a workspace consisting of individual time points and channels.
Users may choose to append existing workspaces to the current one in order to concatenate additional time points. This facilitates the construction of time sequences from otherwise independent image stacks, and is especially useful when working with large time sequences.
Input seeds: Input seeds for the segmentation algorithm may be loaded if available, or they can be added by the user when working on the segmentation. Furthermore, if the dataset represents a time sequence, such input seeds can be propagated from neighboring time points.
Interactive segmentation: Interactions include modifying, deleting, and adding segments. We introduce a cellness confidence measure that models the segmented cells’ features and reas of uncertainty for the user’s attention. Additionally, the interactive segmentation tool suggests corrections for the user to accept or reject.
High-throughput analysis: CellECT provides the option of running the segmentation tool in a non-interactive mode on multiple time points. This option is useful for high-throughput analyses where results are needed quickly with little human intervention. Further, an option to propagate the segmentation to neighboring time points is available. This transfers seed points from one time point to the next as well as propagates a bias for segment shapes. The user has the option to correct the segmentation results in the interactive mode.
Analysis tool: CellECT provides an analysis module which runs on a selected subset of time points for which segmentation is available. Changes in local and global statistics of various cell measurements over time are tracked. The user has the ability to select regions of interest in order to observe cell behavior. Furthermore, automated clustering can be performed to categorize similar cells.
Exporting results: The segmentation results are available in various formats: slice by slice TIFF files, MAT-file format, and polygon contours in XML files. The analysis and measurements computed for each segmented cell are available in XML format.
Interactive segmentation
The method starts out with the membrane/cell wall channel of the original microscopy volume, V
t
, at a given time point t, and a point cloud of initialization seeds (nuclei) associated with this volume,  . These points, if available, may be imported from an external nuclei detector such as described by [10], or randomly distributed throughout the volume, or propagated from the segmentation available at a neighboring time point.
. These points, if available, may be imported from an external nuclei detector such as described by [10], or randomly distributed throughout the volume, or propagated from the segmentation available at a neighboring time point.
In this section, we limit the discussion to the interactive segmentation, analysis and confidence evaluation of a single volume at a given time t and use the simplified notation of V and  for V
t
and
for V
t
and  respectively. Though this is not a limitation on the overall methodology, the notation in the analysis that follows refers to a single time point t.
respectively. Though this is not a limitation on the overall methodology, the notation in the analysis that follows refers to a single time point t.
At each iteration the user contributes a set of guidance marker points, \(\mathcal {P}^{i}\), where each marker point x
p
is described by its spatial coordinates within the given volume: x
p
=[x,y,z]. The initial input seed points,  , along with guidance marker points resulting from subsequent user interaction are maintained in a graph \(G^{i} = \left \{\mathcal {V}^{i}, \mathcal {E}^{i}\right \}\), where i is the user interaction iteration index, and
, along with guidance marker points resulting from subsequent user interaction are maintained in a graph \(G^{i} = \left \{\mathcal {V}^{i}, \mathcal {E}^{i}\right \}\), where i is the user interaction iteration index, and  . Each seed \(\mathbf {x}_{p} \in \mathcal {V}^{i}\) is associated with a segment in the final segmentation, and each segment is described by at least one seed. The index of the segment associated with a seed is given by Seg(x
p
)=k. There exists an edge in the graph \(e_{\textit {pq}} \in \mathcal {E}^{i}\) if Seg(x
p
)=Seg(x
q
). In summary, the nodes in the graph represent seed points, and the edges in the graph model the membership of seeds to disjoint subsets (connected components in the graph). Each disjoint subset contains the seeds (nuclei and user guidance markers) associated with one segment.
. Each seed \(\mathbf {x}_{p} \in \mathcal {V}^{i}\) is associated with a segment in the final segmentation, and each segment is described by at least one seed. The index of the segment associated with a seed is given by Seg(x
p
)=k. There exists an edge in the graph \(e_{\textit {pq}} \in \mathcal {E}^{i}\) if Seg(x
p
)=Seg(x
q
). In summary, the nodes in the graph represent seed points, and the edges in the graph model the membership of seeds to disjoint subsets (connected components in the graph). Each disjoint subset contains the seeds (nuclei and user guidance markers) associated with one segment.
User interactions model such actions as “merge two segments”, “modify a segment”, “delete a segment”, and “add a new segment”, by manipulating the graph of input markers G i. For example, to modify a segment additional guidance markers (seeds) can be provided and associated with the given segment. To delete a segment, the seeds associated with it are eliminated. The seeds (nuclei and user markers) and their subset membership are efficiently maintained as a disjoint set data structure [11], permitting find and union operations with computational complexity \(O(log \mathcal {V}^{i})\) and constant amortized complexity. The graph which is actually is actually a forest of tree graphs, is implemented internally using two arrays.
At each iteration i, a seeded Watershed segmentation method [12] takes as input a membrane (or cell wall) volume V and a spatial arrangement of seed markers along with their subset membership relationship as modeled by the configuration of graph G i. This results in a segmentation label map, S i, in which every pixel is assigned a label corresponding to a segmented cell or the image background.
The segmentation label map S i is evaluated by computing the cellness metric of each segment (detailed in Section “Learning a cellness metric”). The cellness metric uses segment measurements to return a confidence value which models the likelihood that the segment correctly represents a segmented cell. The cellness metric is used to highlight regions of uncertainty for the user’s attention.
Once the segmentation label map and the cellness metric evaluation of each segment are computed, the user may once again modify the segmentation. The process repeats until the user is satisfied with the results.
Watershed markers
User guidance seeds play a vital role in initializing the seeded watershed segmentation algorithm. The watershed algorithm is a segmentation algorithm which flood fills the image space starting from input markers, interpreting the image as a topological relief, where pixel intensity is analogous to altitude. The input markers may be a single point per segment or a series of strokes through the segment volume. CellECT enables users to interact with the watershed segmentation algorithm by manipulating the input markers through guidance seeds.
Each input seed point, whether detected using a nuclei detector or manually marked, translates to one input marker for the segmentation algorithm, which flood fills its neighborhood to form a segment. In order to modify a segment, the user places additional guidance seeds at each iteration i, which are maintained in the graph G i.
When multiple guidance seeds are provided for the same segment, CellECT combines them to form stroke markers in the 3D space. This is done by computing the geodesic shortest path in the image space between one central seed and all other seeds describing the same segment k. The central seed, given by image space coordinates \(\mathbf {x}_{k}^{c}\), is picked to minimize the Euclidean distance to the mean location \(\overline {\mathbf {x}}\) of all the seeds \(\mathbf {x} \in \mathcal {V}^{i}_{k}\) describing a segment k:
The geodesic shortest path between the seed \(\mathbf {x}_{k}^{c}\) and the other seeds pertaining to segment k, \(\mathcal {V}^{i}_{k} \setminus \left \{\mathbf {x}_{k}^{c}\right \}\), connects all seeds describing to segment k. The shortest path is computed using gradient descent over the distance function \(\mathcal {D}\). \(\mathcal {D}\) can be obtained by solving the Eikonal Eq. 2 using the Fast Marching algorithm introduced in [13].
where P(·) is the speed of the propagating front, embedded in a higher dimensional level set function. If the speed P is constant, the resulting distance function \(\mathcal {D}\) can be seen as the distance function to the starting point, \(\mathbf {x}_{k}^{c}\). Gradient descent on this distance function returns the shortest path in geodesic image space from each point in \(\mathcal {V}^{i}_{k} \setminus \{\mathbf {x}_{k}^{c}\}\) to the starting point \(\mathbf {x}_{k}^{c}\).
The resulting shortest paths connecting the guidance markers for each segment together with the point cloud of input seeds are given as input markers to the Watershed algorithm. Figure 3 shows an example of such input guidance markers, the resulting segmentation and the original image. Guidance markers pertaining to the same cell must be connected to each other in order to prevent the segmentation of one to result in disjoint fragments. The geodesic shortest path connects seeds through curved paths without crossing cell boundaries.

User markers. a Seeds (in black), and the shortest path through the geodesic image space between identically-labelled seeds. These seeds and paths are given as initialization points or strokes to a seeded Watershed algorithm. b An x−y and an x−z plane through the resulting segmentation. c the corresponding planes through the original image
Segment features
Once the segmentation is computed using the input markers described in Section “Watershed markers”, several properties are calculated for each segment at every iteration. These features are further used in calculating the cellness metric (Section “Learning a cellness metric”) to guide user interactions and for computing cell statistics (Section “ CellECT recommendations, segment propagation and cell analysis”).
Segment border features
capture the implicit assumption that the membrane or cell wall channels are expected to have higher intensity signal than the cell interiors.
1) Border to interior intensity ratio is given by \(\frac {\overline {\mathcal {B}_{k}}} {\overline {\mathcal {S}_{k}}}\), where \(\overline {\mathcal {B}_{k}}\) represents the average intensity along the border of segment k and \(\overline {\mathcal {S}_{k}}\) represents the average signal intensity within segment k.
2) Distance between border and interior intensity histograms. This is computed using the Earth Mover’s Distance [14] and evaluates whether there is any membrane signal present in the segment interior.
Position properties
describe the segment’s position in the image space and relative to the specimen’s coordinate system, as explained below.
3) Segment centroid. The centroid coordinates are given with respect to the image origin, rescaled according to the image resolution in each dimension, and is equivalent to the center of mass of the segment.
4) Centroid distance from specimen surface. The minimum Euclidean distance of each segment’s centroid to the specimen’s outer boundary, scaled by the image resolution, is computed. This is given by \( D^{\mathcal {B}}_{\mathcal {B}_{k}}\), where \(\mathcal {B}\) is the boundary (surface) of the specimen in the microscopy volume, \(\mathcal {B}_{k}\) is the boundary of segment k, and the distance transform in Eq. 3 denotes the minimum distance of every point x in set A to the points in set B.
5) Best fit line. A line is fit through the voxel coordinates of segment k using the algorithm of [15], which is based on the M-estimator technique that iteratively fits the line using the weighted least-squares algorithm. The resulting feature consists of a 6-element vector containing a normalized unit vector collinear to the line and a point on the line, in the image coordinate system.
6) Segment position along the anterior-posterior (AP) axis. The AP axis is a set of points \(\mathcal {S}_{\textit {AP}}\) obtained by interpolating a list of consecutive marker points in the image space given by the user, which traverses the specimen from the anterior end to the posterior end. The projection of the centroid of segment k, c k is given by the point on the AP axis with the smallest Euclidean distance to c k . This is given by Eq. 4. Next, the position along the AP axis is calculated as the percentage along the axis starting from the first anterior point to the last posterior point.
7) Segment angle with the AP axis. For every segment k, the unit tangent to the AP axis at the projection point \(\mathbf {c}_{k}^{AP}\) is used to compute the smallest angle with the best fit line unit vector.
8) Segment distance to AP axis. This is given by \(||\mathbf {c}_{k}^{AP} - \mathbf {c}_{k}||\).
Shape and size properties
characterize the 3-D shape of each segment.
9) Segment volume: is given by the voxel count of the segment scaled by the image resolution, \(V_{k} = |\mathcal {S}_{k}| \cdot \mu _{x} \mu _{y} \mu _{z}\), where \(|\mathcal {S}_{k}|\) is the cardinality of the set of voxels occupied by segment k, and μ x , μ y and μ z represent the image resolution scale factor in each dimension.
10) Distance of segment border to segment centroid: This feature computes the histogram of the set of distances between the voxels on the segment border and the segment centroid, \({D_{k}^{c}} = \{ ||\mathbf {x} - \mathbf {c}_{k}|| \cdot \mu | \mathbf {x} \in \mathcal {B}_{k}\}\), where D is the distance function defined in Eq. 3. Here, \(\mu = \frac {1}{\underset {\textbf {x} \in \mathcal {B}_{k}}{\text {max}} ||\mathbf {x} - \mathbf {c}_{k}||}\) is a scale factor such that the maximum element in the set is 1.
11) Sphericity: The radius of a sphere with the same volume as segment k is given by \(r_{k} = \sqrt [3]{\frac {3 \cdot |\mathcal {B}_{k}|}{4 \cdot \pi }}\). The ratio of the surface area of this sphere to the surface area \(\mathcal {B}_{k}\) of segment k is stored as a feature and indicates how much the segment shape deviates from a sphere, and is given by \(\frac {|\mathcal {S}_{k}|}{4\pi \cdot {r_{k}^{2}}}\).
12) Squareness: is given by the ratio of the segment volume and the volume of the minimum enclosing bounding box: \( \frac {V_{k}}{V^{\text {box}}_{k}}\). The minimum oriented bounding box is obtained from the projection extremities of each segment along each of the three principal axes.
13) Cylindricity: This metric evaluates the segment’s deviation from a cylinder. The volume \(V^{\text {cyl}}_{k}\) of the minimum enclosing cylinder oriented along the principal axes of segment k. The lowest \( \frac {V_{k}}{V^{\text {cyl}}_{k}}\) ratio of the three enclosing cylinders represents the cylindricity score.
14) Convexity: The deviation of the segment shape from a convex form is measured as the ratio of the segment volume to the convex hull volume: \(\frac {V_{k}}{V_{k}^{\text {hull}}}\).
15) Entropy: is a measure of compactness and is calculated using the eigen values obtained from principal component analysis, as in [16].
16) Elongation: Similar to [16], the elongation is given by the ratio of the largest eigenvalue to the midium eigenvalue: \(\frac {\lambda _{\text {max}}}{\lambda _{\text {med}}}\).
17) Flatness: Similar to [16], the elongation is given by the ratio of the medium eigenvalue to the smallest eigenvalue: \(\frac {\lambda _{\text {med}}}{\lambda _{\text {min}}}\).
Learning a cellness metric
A novel CellECT feature is its ability to highlight uncertain segmentation results to the user. A confidence metric, called cellness, is constructed for each dataset based on a continuous learning framework that models various cell features described above. The model is continuously updated based on user interactions.
Expected segment characteristics
Figure 4 shows several examples of incorrect segments. We argue that a correctly segmented cell has at least one of these distinct characteristics: (1) the boundary of the segment has higher intensity than the interior, (2) the boundary between a cell and each of its neighboring segments has high intensity, (3) cells are mostly convex, and (4) cells are on average similar to their neighbors. Inconsistent intensity along the segment boundary is often an indicator of a segment which does not adhere to membrane or cell wall staining, for example the result of over-segmentation. The cell neighbor similarity expectation is motivated by the fact that cells usually develop in compact tissues which maintain consistent appearance. Shape and size features were chosen for cell similarity since these are the cues humans typically look for to identify incorrectly segmented cells. Viewing these measurements outside their local context and independent of each other may not be effective when working with multiple tissues, whose cells differ in appearance. Therefore, in addition to quantifying each of these properties, the cellness metric adapts to user feedback. Metrics (1)-(3) are obtained as explained in Section “Segment features”, and denoted \({s_{k}^{1}}\), \({s_{k}^{2}}\) and \({s_{k}^{3}}\). Metric (4), \({s_{k}^{4}}\), is computed by calculating the average distance in feature space between segment k and each of its neighbors j, as in Eq. (5). Here, f k (f j ) is the feature vector obtained by concatenating the values of volume, sphericity, flatness, and elongation obtained as described in Section “Segment features” and normalized zero mean and unit standard deviation. The scale factor c brings \({s_{k}^{4}}\) to range between 0 and 1. Thus, the values of \({s^{1}_{k}}\) through \({s^{4}_{k}}\) range between 0 and 1, where large values are obtained by segments which meet the characteristics (1)-(4) above.

Example of problematic segments. A correct segment exhibits one or more of these characteristics: the boundary signal intensity as stronger than the interior, the common boundary with a neighboring cell has high intensity, the segment shape is almost convex and the segment’s shape is similar to that of its neighbors. Incorrect segments indicated by arrows: a segment is far from convex, b signal intensity in the membrane channel is not high on the segment border, c signal intensity in the membrane channel is too high in the segment interior, d–e segments are not similar to their surroundings
Learning region characteristics from positive user examples
User feedback is of two types: a problematic segment may be corrected, or a segment may be marked as correct. Segments marked as correct provide information about the expected segment measurements in their neighborhood. Segments in the neighborhood of positive user feedback are compared against the user examples under the spatial homogeneity assumption and scored accordingly.
Each segment is modeled as a node in a graph and edges are introduced between nodes representing neighboring cells. Given a set of segments marked by the user as correct, the task is to disseminate this confidence credit to other segments in the graph, based on their similarity to and distance from the correct segments, as shown in Fig. 5 a-b. We consider the simplified problem of disseminating the credit from each correct segment to every node in the graph along the optimal path.

a Segments represented as nodes in a graph. Edges connect neighboring segment. Two segments (p 1 and p 2) marked by the user as correct. b Disseminate credit from the correct segments to other segments in the graph, based on similarity within the neighborhood. c Confidence credit disseminated from segment p 1 to all other segments along the path of highest similarity. d Credit disseminated from p 2
The optimal path from a segment k to a segment p labeled as correct is the path which maximizes the probability that every segment along the path is correct. This can be written as in Eq. 6, in which a path through the graph between nodes (segments) k and p is defined by the set of vertexes \({P_{k}^{p}} = \left \{v_{1}, v_{2},.. v_{n} \right \}\), where v 1=p, v n =k and P(v 1)=1.
We model the probability P(v i |v i+1,v 1) as the pairwise similarity between the two neighboring nodes v i and v i+1, which is independent of v 1. This can be expressed as P(v i |v i+1,v 1)=P(v i |v i+1)=d i,i+1. The similarity measure d i,i+1 is given by 1−||f i −f i+1||·c, where f i and f i+1 are the feature vectors defined earlier and c is the scaling factor in Eq. (5). Therefore, the goal is to obtain the path that maximizes the pairwise similarity between segment k and the correctly labeled segment p, i.e., \(\mathcal {A}\left ({P_{k}^{p}}\right) = \prod \limits _{i=1}^{n} d_{i,i+1}\).
To find the above optimal path, we note that:
where w ij =− logd ij are non-negative weights. Therefore, the method described by [17] can be used to obtain the shortest path from a correct node p to every node in the graph, as shown in Fig. 5 c-d. Finally, the confidence credit obtained by a segment k from a set of correctly labeled segments p is given by:
Hence, \({s_{k}^{5}}\) quantifies the confidence that segment k is correct assuming the user input and the knowledge about its neighborhood, in terms of similarity metrics.
An example of the effects of this metric are shown in Fig. 6 b.

Cellness metric example. a Slice through the original confocal microscopy image. b Propagation of confidence from segments marked as correct (indicated by arrows) to similar neighbors. c Color coded cellness metric. d Reconstructed cross section in the x−z plane of the cell with low cellness metric indicated by arrow in panel. This segment appears correct in the view from panel C however it has a low cellness score. e Error in the segmentation, indicated by arrow, observable in the cross section (the segment leaks into the cell below). The cellness metric helped identify this error in segmentation
Metric learning from positive and negative user examples
The cellness metric adapts to user feedback. Segments which are marked as correct are used as positive examples, while segments that are corrected through user interactions are used as negative examples. We use the semi-supervised learning approach of [18] which is designed to work well if given a very small number of labeled samples, together with a large number of unlabeled samples. The feature space consists of \(\left ({s_{k}^{1}},{s_{k}^{2}},{s_{k}^{3}},{s_{k}^{4}}\right)\) tuples, and two class labels are considered: correct segment and incorrect segments. The output of the classifier is the probability that each sample belongs to the correct class, which we denote \({s_{k}^{6}}\).
A correct segment will obtain high values for metrics \({s_{k}^{1}}\) through \({s_{k}^{5}}\) and, if the classifier is trained well, the segment will obtain a high value for \({s^{6}_{k}}\). The cellness metric combines \({s_{k}^{1}}\) through \({s_{k}^{6}}\), giving more weight to either the mean of \({s_{k}^{1}}\) through \({s_{k}^{5}}\) or to the result of \({s_{k}^{6}}\), according to Eq. 9. The average of \({s_{k}^{1}}\) through \({s_{k}^{5}}\) gives equal weight to each of the five sub-metrics. The output of the classifier produces a more complex decision boundary tailored to the dataset. This score is reliable if a balanced number of positive and negative training samples are available, and sufficient training examples are available. Thus, the weight factor ν ranges between 0 and 1 and evaluates to a higher values for a larger number of training examples and a balanced number of positive and negative samples.
Figure 6 c shows an example of the cellness confidence evaluation in a 3D volume of the ascidian P. mammillata. The colors in Fig. 6-c represent the degree of confidence (low to high: red-yellow-green). The red cell indicated by an arrow appears correctly segmented. However the reconstructed cross section in x−z reveals an error in the segmentation, as observed in Fig. 6 d-e.
CellECT recommendations, segment propagation and cell analysis
In this section we discuss several features in CellECT, such as correction recommendations for the user, segmentation propagation to neighboring time points, and analysis tools for segmented volumes.
Recommendations
CellECT identifies problematic segments for the users to validate. Examples of such segments include spurious boundaries due to weak signal to noise ratio or dividing cells for which a nuclei detector may have discovered more than one nucleus. The mean intensity on the common boundary \(\mathcal {B}_{\textit {kj}}\) of segment k and its neighbor j is given by \( \overline {\mathcal {B}_{\textit {kj}}}\). A merging score given by \(\overline {\mathcal {B}_{\textit {kj}}} \cdot \frac {|\mathcal {B}_{\textit {kj}}|} { |\mathcal {B}_{k}|} \) is computed for every pair of neighboring segments. Pairs of segments are suggested to the user for merging or deletion in the increasing order of their scores.
Segment propagation
In order to facilitate high-throughput analysis CellECT allows users to propagate the segmentation results from one time point to the next. A simple approach is to transfer the background location and an interior point of each segment as seed points to the next (or previous) consecutive time point.
The segment inner point is given by the maximum of the distance transform (Eq. 3) from the segment boundary applied in the interior of the segment. Thus, the segment inner point is given by \(\text {arg}\max \limits _{\mathbf {x} \in S_{k}} D_{\mathbf {x}}^{\mathcal {B}_{k}}\), where \(\mathcal {S}_{k}\) denotes the interior of segment k and \(\mathcal {B}_{k}\) denotes the border of segment k.
The new seeds will serve as input seeds to the new segmentation. In the event of errors in the resulting segmentation, the user can correct the segmentation by placing additional guidance seeds. Segmentations may be propagated using the interactive segmentation tool, or using the segmentation tool in non-interactive mode analysing a batch of time points together.
Cell analysis
CellECT includes an analysis tool, applicable to the multiple time-point segmentation results computed in the fashion described earlier. This tool can compute multiple local/global statistics as well as keep track of their changes over a subset of time points. Regions of interest may be selected for analysis by constraining position coordinates relative to the specimen.
Additionally, CellECT has a clustering module which implements K-means clustering algorithm. This module enables the user to cluster cells in a given volume based on similarity in a user-defined feature space. The user can select one or more features (Section “Segment features”) and specify the number of desired clusters for grouping cells. These parameters can be adapted to the data, and the resulting clusters can be visualized in the segmented volume. A number of group statistics (e.g. inter/intra-group variance, average cluster center distance etc.) are computed per volume over the time-series.
Results and Discussion
Ascidians are used in the study of animal morphogenesis due to their small size, simple and compact embryo, and its similarity in early development to vertebrates. The Smith lab at UCSB uses microscopy volumes of ascidians for quantitative analysis in morphogenesis research [19, 20].
Two 3D+t datasets of the ascidian P. mammalitta are analyzed using CellECT. The first dataset, Ascidian-18, is a confocal microscopy time series which consists of 18 time points (26 slices per volume), from stage 15 to stage 21 [21] with membrane and nuclei channels. This dataset starts out with approximately 300 cells which develop into 500 cells. The second dataset, Ascidian-192, is a SPIM time series which consists of 192 time points (197 slices per volume), from stage 6 to stage 19, also with membrane and nuclei channels. This dataset starts out with 32 cells which develop into almost 1000 cells.
Additionally, a third case study from a different application is considered: The leaf and cotyledon epidermal cells of dicot plants are highly interdigitated with a jigsaw-puzzle piece shape. Using Arabisopsis as a model, it has been shown that the growth properties of the epidermis influence the size and shape of the organ [22]. Therefore, understanding how the growth properties of the cell relate to organ form is an important biological question. Historically, measurements of these cells have been done by manually segmenting each cell [23–26], a highly time-consuming procedure, but recently there has been a push for a more automated approach [27]. The A. thaliana dataset consists of 112 individual cells from 2D confocal microscopy slices which were segmented using CellECT.
Ascidian P. mammalitta - 18
The method in [10] is used to detect nuclei. Segmentation results corresponding to the first and last time points are presented in Fig. 7 a-d. CellECT’s analysis module is used to isolate several regions of interest and observe changes over the development of the embryo. This datasets captures the development of the embryo up to tailbud stage 21 [21]. The following regions of interest are relevant at the tailbud stage: the notochord tissue forms in the posterior half of the embryo along the AP axis; muscle cells develop surrounding the notochord tissue; the epidermis develops at the surface of the embryo; finally, the cells in the dorsal half of the embryo, below the epidermis, represent the neural tube. These four regions of interest are marked by restricting the spatial properties computed in Section “Segment features”. Figure 7 f-g shows the spatial arrangement of the cell nuclei color coded by their membership to one of these regions of interest.

Ascidian-18 dataset: a–d original slice and respective segmentation for t=0 (stage 15) and t=17 (stage 21) (E) Clustering of cells with similar properties identifies tissues f – g Nuclei at t=0 and t=17 in each of the four regions of interest: notochord (yellow), muscle (blue), endoderm and neural tube (red), epidermis (green). h–i Average cell measurments over time per region of interest: h volume i flatness
Statistics over various cell measurements in each of these regions of interest are computed. For example, Fig. 7 h-i shows the average volume and cell flatness over the 18 time points in each region. As expected from the known development of the ascidian tailbud, notochord cells become mostly flat, followed by muscle cells. Also, as a result of cell divisions the average volume over time decreases, while muscle cells and endodermal cells maintain the highest volume. These measurements confirm the expected developmental behavior, suggesting that the segmentation label maps resulting from CellECT are accurate.
Additionally, segments are clustered in feature space in order to group similar cells using K-means clustering algorithm [28]. The inter-cluster distance was computed for every time point, and an increasing trend was observed (from 2.6 units in the normalized feature space to 3.6 units). This is due to the fact that cells specialize as they form tissues. These measurements meet the expected behavior, suggesting that the segmentation label maps resulting from CellECT are reliable. An example of such clustering is shown in Fig. 7e, where the color coding marks each of the four clusters.
Ascidian P. mammalitta - 192
The nuclei detector of [10] did not perform well on this dataset due to artifacts such as those in Fig. 1 a, f. Only about 500 out of the approximately 900 cells in the last time point were detected. CellECT’s interactive segmentation tool is used to correct and propagate the segmentation results. The interactive recommendation feature in the segmentation module was used to quickly identify the dividing cells. Figure 8 a-d shows a slice through the first and the last time points of the dataset and their respective segmentation.

Ascidian-192 dataset: a–d original slice and respective segmentation for t=0 (stage 6) and t=192 (stage 19) e–g: Superimposed histograms of segment measurments, color coded by time point (from blue to red): e volume, b sphericity, c entropy
This dataset starts out with 32 cells in the first time point and ends with approximately 900 cells in the last time point. Using CellECT analysis module various measurements are computed over the time sequence. Figure 8 e-g shows three of these measurments over time: the histogram of volume of cells at each time point, the histogram of sphericity values over time and the histogram of entropy values over time. Each histogram is color coded by the time point it represents. As expected, cells exhibit lower surface area in the later time points. This is a result of cell division resulting in many more cells occupying approximately the same total volume as the early time points. Similarly, cells in later time points exhibit lower entropy, suggesting that cell shape become more compact over time.
Arabidopsis pavement cells
Images were collected using a scanning confocal microscope with a 40X-oil immersion objective [29]. This dataset lacks a nuclear marker to automatically identify each cell. Cell identification is assigned manually and segmentation is based on fluorescent signal of the lipophilic dye FM4-64 which labels cell periphery. Cell segmentation is restricted to pavement cells that are completely contained within the image field. The same cells are also segmented manually and morphometric measures (area, perimeter and circularity) are obtained on both sets. These measures are compared against each other to determine the quality of the segmentation in Section “Quantitative evaluation of segmentation quality using A. thaliana slices”. Small symmetrical cells associated with the stomatal lineage are also accurately segmented using CellECT, but are not included in the morphometric analyses.
CellECT reduced the necessary time for cell segmentation by a factor of ten when compared to manual methods. Two examples of confocal slices and the respective segmentations are shown in Fig. 9.

A. thaliana pavement cells: Left column: Original slices, Middle: Segmentations overlaid on original slices, Right: Segmentation label maps
Analysis
Next, we evaluate the quality of the segmentations obtained using CellECT and the efficiency in using the cellness metric.
Quantitative evaluation of segmentation quality using A. thaliana slices
The segmentations obtained in Section “Arabidopsis pavement cells” are compared against manually obtained segmentations in order to evaluate the segmentation quality. XY-coordinates were extracted from CellECT’s results and imported as a ROI into the scientific image viewing software, ImageJ [30]. Manual segmentation was conducted on the same cells segmented by CellECT using the polygon selection tool in ImageJ with the spline function active.
A total of 112 pavement cells were segmented from three different plants all taken at the same developmental stage. The majority of the cells in the field were accurately segmented, and an average of 14 pavement cells from 5 day after germination (DAG) cotyledons were obtained from an image field of 308 × 308 μm. Measurements of cell area, perimeter, and circularity (unitless shape descriptor), which are commonly used for pavement cell analyses, were taken from cell outlines that were manually segmented and those obtained using CellECT. Cell population from each image field were tested for normal distribution and analyzed using either Student’s t-test for populations with normal distribution or Mann-Whitney test for populations with non-normal distributions. A p-value less than 0.05 indicates a statistical significant difference between parameter outputs from CellECT and manual segmentation Table 1.
Quantitative evaluation of segmentation quality and CellECT’s segmentation propagation feature using the Ascidian-192 dataset
In this section we compare the segmentations obtained using CellECT’s workflow against manually generated 3D ground truth for a subset of cells. We compare different approaches in order to determine the benefits of the interactive segmentation feature.
To demonstrate the utility of propagation over consecutive time points, a set of fifty cells spanning over five consecutive time points was manually traced in 3D. Ten cells per time point were selected for the last five time points of the Ascidian-192 dataset and ground truth segmentation was generated. This set contains cells from different tissues, as well as special cases such as dividing cells. The metric used for comparison is the F-measure which is a volume based error metric defined as
where P and R are the precision and recall of the corresponding ground truth volume.
In order to evaluate the utility of the segmentation tool, four different segmentation approaches were compared, and the F-measure was evaluated for every cell for which ground truth is available. The F-measure corresponding to the fifty segmented cells is shown in Fig. 10 a, where the x-axis corresponds to the index of the fifty cells. Cells 1–10, 11–20, 21–30, 31–40 and 41–50 correspond to time points 188, 189, 190, 191 and 192, respectively. Four different approaches to the segmentation of these five consecutive volumes were explored:
-
1.
First, the volumes were segmented by initializing the algorithm with the output of the nuclei detector of [10]. This dataset is particularly challenging for a nuclei detection algorithm as shown in Fig. 1 a and Fig. 1 f. As seen in Fig. 10 a this initialization results in the incorrect segmentation of several cells. Using the interactive segmentation tool, the initialization errors can be corrected and subsequently propagated to neighboring time points, which in turn may be corrected in the event of any additional mistakes. These workflows are explored next.
Fig. 10 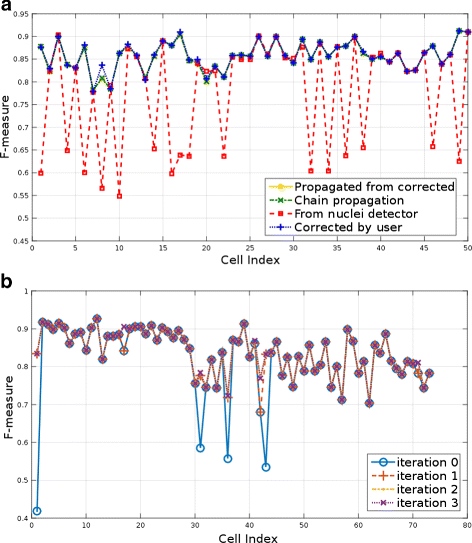
Quantitative evaluation of the segmentation: a F-measure for the segmentation of fifty randomly selected cells from the last five time points of the Ascidian-192 dataset. Four segmentation results are compared: (1) nuclei detector initialization, (2) propagation of corrected volumes, (3) chain propagation without corrections, (4) propagated and corrected volumes. b F-measure for four approaches to the segmentation of time points 188–192 from the Ascidian-192 dataset
-
2.
The second approach consists of correcting the last time point (cells with index 41–50) and propagating this corrected segmentation to the previous four time points (cells 1–40). Thus a chain of propagated segmentations is obtained without any user intervention, except for the last time point.
-
3.
The third approach measures the quality in segmentation when every propagated volume is corrected for potential errors. This approach evaluated the quality of a segmentation propagated from a corrected result and before any additional human interaction.
-
4.
Finally, the fourth approach measures the quality of segmentation when propagating from a corrected volume and after correcting any resulting errors.

As seen in Fig. 10 a, there are little differences in the scores of the three propagation approaches and they compare favorably to the results without any human intervention. Therefore, propagating a corrected volume reduces the need for human intervention. The average F-measure for each of the approaches is listed in Table 2. In conclusion, the interactive segmentation tool coupled with the propagation feature enables segmentation with reduced human interaction of large time sequences for which automated methods often have difficulty. For example, in Fig. 10 a, several cells obtained scores as low as 55 % with automated nuclei detection, but instead, using CellECT’s features, a correct segmentation can be propagated to subsequent time points. As a result, F-scores above 80 % can be obtained without any additional user interaction.
Next, we use 73 cells from the first time point of the Ascidian-18 dataset for which manually traced 3D ground truth is provided. We observe improvements in segmentation over multiple user feedback iterations using the F-measure for each cell. As seen in Fig. 10 b the problematic cells are corrected and a satisfactory segmentation is obtained in four iterations. The average F-measure at each iteration is listed in Table 3, where the first iteration starts out with the output from the nuclei detection algorithm. The most significant corrections are made in the early iterations.
Quantitative evaluation of cellness metric performance using Ascidian-18 dataset
In this section we investigate if the cellness metric can effectively identify incorrect segments. The following experiments are performed using segmentations from the Ascidian-18 dataset. In the first experiment we compare the cellness metric score for two sets of cells which are manually annotated into one of two classes: “Correct” and “Incorrect”. An effective cellness metric is expected to show a distinct separation between the two classes. This experiment is performed on five time points of the dataset, where approximately 15–20 cells of each class are selected at each time point. In a second experiment, ten “Correct” and ten “Incorrect” cells are selected from the final time point, for which each component of the cellness score is observed.
In the first experiment, we manually identify 15–20 correctly segmented cells from each of the time points t=0,4,8,12,17. Similarly 15–20 incorrectly segmented cells are manually selected from each of these five time points. The cellness metric for the cells in these two categories is plotted in sorted order in Fig. 11. The two categories of cells separate well for every time point. The average cellness metric for the “Correct” and “Incorrect” groups for these time points is listed in Table 4. As expected, “Correct” cells consistently obtain a higher cellness metric than the set of “Incorrect” cells in each time point.

Cellness metric for correct and incorrect cells. Cellness metric in sorted order for hand picked cells in two categories, “Correct” and “Incorrect”, over five time points (t=0,4,8,12,17) of the Ascidian-18 dataset. The two classes of cells separate well for the different time points
Next, in the second experiment, we examine the contribution of each component of the cellness metric. Ten “Correct” cells and ten “Incorrect” cells are selected from the last time point of the dataset. Fig. 12 shows the contribution of each cellness score for every cell of the “Correct” and “Incorrect” categories: \({s_{k}^{1}}\), \({s_{k}^{2}}\), \({s_{k}^{3}}\), \({s_{k}^{4}}\) and \({s_{k}^{5}}\) and c e l l n e s s(k) described in Section “Learning a cellness metric”. In addition, Fig. 13 and Table 5 show the average score for each cellness component over the ten cells of each category. This experiment demonstrates that the cellness metric effectively captures cues which indicate the quality of the segmentation.

Cellness metric components. Score of each cellness metric component over ten “Correct” and ten “Incorrect” cells from t=17 of the Ascidian-18 dataset, and the combined cellness score

Average cellness metric components. Average scores over the ten cells in each class for each cellness metric component, the average of all components, and the combined score (cellness). “Correct” cells obtain a higher cellness score than “Incorrect” cells
Conclusion
We introduced a software for the interactive segmentation and analysis of 3D+t membrane or cell wall image datasets. CellECT enables users to create and interact with the segmentation of images containing cell boundary information by adding, deleting, or modifying segments. An adaptive confidence metric (cellness metric) helps identify areas of uncertain segmentation. The algorithm is able to identify spurious boundaries and suggest corrections. Segmentation results can be propagated to neighboring time points. Once segmentation is obtained for multiple consecutive time points the analysis tool displays statistics over time and allows the user to focus on regions of interest.
We demonstrated to utility of this framework by quantitatively evaluating the quality of segmentations and the efficiency of the cellness metric. Case study analysis was performed on three datasets: a time series of 18 volumes of the of the Ascidian P. mammillata, a time series of 192 volumes of the same species, and a set of 112 cells from 8 confocal slices of A. thaliana pavement cells. Cells from different time points of the two P. mammillata datasets were compared against manually segmented cells. Additionally, the efficiency of CellECT’s segmentation propagation feature and the utility of the cellness metric were demonstrated in quantitative analysis. In the case of the A. thaliana, CellECT reduced the segmentation time by a factor of 10 when compared to manual methods without reducing the quality of segmentations. No statistical significant differences were found between CellECT coordinates and manually extracted cells in the parameters of area, perimeter, and circularity.
Future work aims at developing a cell lineage reconstruction module. An integrated framework which jointly detects nuclei, computes cell segmentation and reconstructs lineage over the time series in a continuously adaptive feedback loop is desired. Additional future plans include the integration with BISQUE, introduced in [31], an online web-based bio-image analysis system which facilitates collaboration among biologists.
Availability of supporting data
CellECT is an open source project available at http://bioimage.ucsb.edu/. Supplementary material is available such as demo video (Additional file 1), detailed results (Additional file 2) and animations (Additional files 3-9).
References
Derde M, Antanas L, De Raedt L, Guiza Grandas F. An interactive learning approach to histology image segmentation. In: Proceedings of the 24th Benelux Conference on Artificial Intelligence: 2012. p. 1–8.
Sommer C, Straehle C, Kothe U, Hamprecht FA. Ilastik: Interactive learning and segmentation toolkit. In: Biomedical Imaging: From Nano to Macro, 2011 IEEE International Symposium On. IEEE: 2011. p. 230–233.
McGuinness K, O’Connor NE. A comparative evaluation of interactive segmentation algorithms. Pattern Recognit. 2010; 43(2):434–44.
Du CJ, Marcello M, Spiller DG, White MR, Bretschneider T. Interactive segmentation of clustered cells via geodesic commute distance and constrained density weighted nyström method. Cytometry Part A. 2010; 77(12):1137–47.
Lin G, Chawla MK, Olson K, Barnes CA, Guzowski JF, Bjornsson C, et al. A multi-model approach to simultaneous segmentation and classification of heterogeneous populations of cell nuclei in 3d confocal microscope images. Cytometry Part A. 2007; 71(9):724–36.
Peng H, Bria A, Zhou Z, Iannello G, Long F. Extensible visualization and analysis for multidimensional images using vaa3d. Nat Protoc. 2014; 9(1):193–208.
Delgado-Gonzalo R, Chenouard N, Unser M. Spline-based deforming ellipsoids for interactive 3d bioimage segmentation. Image Process. IEEE Trans. 2013; 22(10):3926–40.
Uzunbas MG, Chen C, Metaxas D. An efficient conditional random field approach for automatic and interactive neuron segmentation. Med Image Anal. 2016; 27:31–44.
Vincent L, Soille P. Watersheds in digital spaces: an efficient algorithm based on immersion simulations. IEEE Trans Pattern Anal Mach Intell. 1991; 13(6):583–98.
Fick RHJ, Fedorov D, Roeder AHK, Manjunath BS. Simultaneous cell tracking and image alignment in 3D CLSM imagery of growing arabidopsis thaliana sepals. In: Biomedical Imaging (ISBI), 2013 IEEE 10th International Symposium on. IEEE: 2013. p. 914–917.
Tarjan RE. Efficiency of a good but not linear set union algorithm. J ACM (JACM). 1975; 22(2):215–25.
Meyer F, Beucher S. Morphological segmentation. J Vis Commun Image Representation. 1990; 1(1):21–46.
Sethian JA. Fast-marching level-set methods for three-dimensional photolithography development. In: SPIE’s 1996 International Symposium on Microlithography. International Society for Optics and Photonics: 1996. p. 262–272.
Rubner Y, Tomasi C, Guibas LJ. The earth mover’s distance as a metric for image retrieval. Int J Comput Vis. 2000; 40(2):99–121.
Bradski G. The OpenCV Library. Dr. Dobb’s Journal of Software Tools. 2000. http://www.drdobbs.com/open-source/the-opencv-library/184404319.
Tassy O, Daian F, Hudson C, Bertrand V, Lemaire P. A quantitative approach to the study of cell shapes and interactions during early chordate embryogenesis. Current Biol. 2006; 16(4):345–58.
Dijkstra EW. A note on two problems in connexion with graphs. Numerische Mathematik. 1959; 1(1):269–71.
Zhou D, Bousquet O, Lal TN, Weston J, Schölkopf B. Learning with local and global consistency. In: Advances in Neural Information Processing Systems, 16(16). MIT Press: 2004. p. 321–328.
Delibaltov DL, Ghosh P, Rodoplu V, Veeman M, Smith W, Manjunath BS. A linear program formulation for the segmentation of ciona membrane volumes. In: Medical Image Computing and Computer-Assisted Intervention–MICCAI 2013. Berlin Heidelberg: Springer: 2013. p. 444–451.
Delibaltov D, Ghosh P, Veeman M, Smith W, Manjunath BS. An automatic feature based model for cell segmentation from confocal microscopy volumes. In: Biomedical Imaging: From Nano to Macro, 2011 IEEE International Symposium On. IEEE: 2011. p. 199–203.
Hotta K, Takahashi H, Erives A, Levine M, Satoh N. Temporal expression patterns of 39 brachyury-downstream genes associated with notochord formation in the ciona intestinalis embryo. Development, Growth Differ. 1999; 41(6):657–64.
Savaldi-Goldstein S, Peto C, Chory J. The epidermis both drives and restricts plant shoot growth. Nature. 2007; 446(7132):199–202.
Andriankaja M, Dhondt S, De Bodt S, Vanhaeren H, Coppens F, De Milde L, et al. Exit from proliferation during leaf development in Arabidopsis thaliana: a not-so-gradual process. Dev Cell. 2012; 22(1):64–78.
Zhang C, Halsey LE, Szymanski DB. The development and geometry of shape change in Arabidopsis thaliana cotyledon pavement cells. BMC Plant Biol. 2011; 11(1):27.
Staff L, Hurd P, Reale L, Seoighe C, Rockwood A, Gehring C. The hidden geometries of the Arabidopsis thaliana epidermis. PloS One. 2012; 7(9):43546.
Elsner J, Michalski M, Kwiatkowska D. Spatiotemporal variation of leaf epidermal cell growth: a quantitative analysis of Arabidopsis thaliana wild-type and triple cyclinD3 mutant plants. Ann Bot. 2012; 109(5):897–910.
Roeder AHK, Chickarmane V, Cunha A, Obara B, Manjunath BS, Meyerowitz EM. Variability in the control of cell division underlies sepal epidermal patterning in Arabidopsis thaliana. PLoS Biol. 2010; 8(5):1000367.
Hartigan JA, Wong MA. Algorithm as 136: A k-means clustering algorithm. J R Stat Soc Series C (Applied Statistics). 1979; 28(1):100–8.
Le J, Mallery EL, Zhang C, Brankle S, Szymanski DB. Arabidopsis BRICK1/HSPC300 Is an Essential WAVE-Complex Subunit that Selectively Stabilizes the Arp2/3 Activator SCAR2. Current Biol. 2006; 16(9):895–901.
Schneider CA, Rasband WS, Eliceiri KW. Nih image to imagej: 25 years of image analysis. Nat Methods. 2012; 9(7):671–5.
Kvilekval K, Fedorov D, Obara B, Singh A, Manjunath B. Bisque: a platform for bioimage analysis and management. Bioinformatics. 2010; 26(4):544–52.
Green RA, Kao HL, Audhya A, Arur S, Mayers J, Fridolfsson HN, et al. A high-resolution c. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell. 2011; 145(3):470–82.
Acknowledgements
The authors would like to thank Dmitry Fedorov for help with nuclei detection. This work was primarily supported by Award No. HD059217 from the National Institutes of Health (DLD, ENS, MK, WS, BSM), and with additional support from National Science Foundation under Award Numbers III-0808772 (DLD, BSM, UG) and MCB/IOS 1249652 (SB, DBS).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DLD designed and carried out research, implemented software and user interface, obtained segmentation on Ascidian-18 and Ascidian-192 datasets and drafted the manuscript. UG obtained cluster analysis results. JK obtained segmentation on Ascidian-192 dataset. MK and ENS collected microscopy data. WS coordinated design requirements for biological applications and reviewed the manuscript. SB obtained A. thaliana image datasets, manually segmented, analyzed its measurements and helped prepared the manuscript. DBS helped to analyze the data and prepare the manuscript. BSM coordinated the overall design, development and evaluations of the image processing methods and helped in preparing the manuscript for publication. All authors read and approved the final manuscript.
Additional files
Additional file 1
CellECT Demo. Video demonstrating the utility of CellECT. (MP4 53900 kb)
Additional file 2
Additional analysis. Additional plots and measurements obtained on Ascidian-18 and Ascidian-192 datasets. (PDF 4850 kb)
Additional file 3
Ascidian-18 middle slice first to last time point. Animation showing the middle slice of the Ascidian-18 dataset from the first to the last time point. (GIF 4640 kb)
Additional file 4
Ascidian-18 middle slice first to last time point. Animation showing the middle slice of the segmentation of the Ascidian-18 dataset from the first to the last time point. (MP4 1790 kb)
Additional file 5
Ascidian-18 first time point. Animation showing the first time point of the Ascidian-18 dataset. (MP4 1270 kb)
Additional file 6
Ascidian-18 first time point segmented. Animation showing the segmentation of the first time point of the Ascidian-18 dataset. (MP4 1290 kb)
Additional file 7
Ascidian-18 last time point. Animation showing the last time point of the Ascidian-18 dataset. (MP4 986 kb)
Additional file 8
Ascidian-18 last time point segmented. Animation showing the segmentation of the last time point of the Ascidian-18 dataset. (MP4 970 kb)
Additional file 9
Cells clustered by features. Animation showing cells clustered by features in the last time point of the Ascidian-18 dataset. (GIF 400 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Delibaltov, D.L., Gaur, U., Kim, J. et al. CellECT: cell evolution capturing tool. BMC Bioinformatics 17, 88 (2016). https://doi.org/10.1186/s12859-016-0927-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-016-0927-7