- Research
- Open access
- Published:
SnoReport 2.0: new features and a refined Support Vector Machine to improve snoRNA identification
BMC Bioinformatics volume 17, Article number: 464 (2016)
Abstract
Background
snoReport uses RNA secondary structure prediction combined with machine learning as the basis to identify the two main classes of small nucleolar RNAs, the box H/ACA snoRNAs and the box C/D snoRNAs. Here, we present snoReport 2.0, which substantially improves and extends in the original method by: extracting new features for both box C/D and H/ACA box snoRNAs; developing a more sophisticated technique in the SVM training phase with recent data from vertebrate organisms and a careful choice of the SVM parameters C and γ; and using updated versions of tools and databases used for the construction of the original version of snoReport. To validate the new version and to demonstrate its improved performance, we tested snoReport 2.0 in different organisms.
Results
Results of the training and test phases of boxes H/ACA and C/D snoRNAs, in both versions of snoReport, are discussed. Validation on real data was performed to evaluate the predictions of snoReport 2.0. Our program was applied to a set of previously annotated sequences, some of them experimentally confirmed, of humans, nematodes, drosophilids, platypus, chickens and leishmania. We significantly improved the predictions for vertebrates, since the training phase used information of these organisms, but H/ACA box snoRNAs identification was improved for the other ones.
Conclusion
We presented snoReport 2.0, to predict H/ACA box and C/D box snoRNAs, an efficient method to find true positives and avoid false positives in vertebrate organisms. H/ACA box snoRNA classifier showed an F-score of 93 % (an improvement of 10 % regarding the previous version), while C/D box snoRNA classifier, an F-Score of 94 % (improvement of 14 %). Besides, both classifiers exhibited performance measures above 90 %. These results show that snoReport 2.0 avoid false positives and false negatives, allowing to predict snoRNAs with high quality. In the validation phase, snoReport 2.0 predicted 67.43 % of vertebrate organisms for both classes. For Nematodes and Drosophilids, 69 % and 76.67 %, for H/ACA box snoRNAs were predicted, respectively, showing that snoReport 2.0 is good to identify snoRNAs in vertebrates and also H/ACA box snoRNAs in invertebrates organisms.
Background
Non-coding RNA genes (ncRNA genes) play important roles in the cell, e.g., structural, catalytic and regulatory functions [1, 2]. The study ncRNAs remains challenging, because laboratory experiments to confirm functions performed by one ncRNA are difficult to be performed, and many distinct computational methods find different results to identify and classify ncRNAs. One key problem is that ncRNA functions are closely associated to their spatial (secondary) structures, which prevents the use of methods to predict protein coding genes based only on their nucleotide sequences (primary structures).
Identification of ncRNAs have been developed for a variety of organisms [3–6], with the objective of constructing sets of different classes of ncRNAs. In particular, snoRNAs [7] are 60 to 300 nt ncRNAs, classified based on their characteristic sequence elements, called boxes, in two main classes: H/ACA box snoRNAs and C/D box snoRNAs. In humans [8], snoRNAs are usually found in intronic regions where, after splicing reaction, they escape from degradation by forming a protein complex [7]. Usually snoRNAs have a short stretch of sequence complementary to target RNAs, like rRNAs, tRNAs and snRNAs, performing chemical modifications on them. C/D box snoRNAs contains fibrillarin that promotes the 2’O-methylation on target RNAs, while H/ACA box snoRNAs contains dyskerin that catalyzes the conversion of uridine to pseudouridine [7, 9].
H/ACA box snoRNA and C/D box snoRNA have distinct secondary structures. H/ACA box snoRNAs are formed by a double hairpin loop structure with two short-single stranded regions containing box H (ANANNA), located between the two hairpins loops, and box ACA (ACA) followed by 3 nt upstream the 3’ end. The hairpin loops have bulges, or recognition loops, which form the antisense element for target RNAs. Normally the first unpaired nucleotide inside the recognition loop is an uridine located 13–16 nt before the H and ACA boxes [7, 10, 11]. Figure 1 shows a schematic secondary structure of H/ACA box snoRNA.

Example of H/ACA box snoRNA
C/D box snoRNAs are formed by two conserved boxes C (RUGAUGA, where R is a purine) and D (CUGA) near their 5’ and 3’ ends, separated by a short stem (3–10 nt). Inside the loop between C and D boxes, usually there are imperfect copies of C and D boxes, called C’ and D’. Normally the antisense element is located 5 nt upstream C’ and D’ boxes. Figure 2 shows a schematic secondary structure of a C/D box snoRNA.

Example of C/D box snoRNA
SnoReport [9] is a tool that identifies the two main classes of snoRNAs in single sequences, using a combination of secondary structure prediction and machine learning. In contrast to previous methods for snoRNA identification (except snoSeeker [10]), snoReport prediction does not use information of putative target sites within ribosomal or spliceosomal RNA (this information can dramatically improve identification sensibility and specificity). However, many orphan snoRNAs have been discovered with the snoReport approach. The targets of orphan snoRNAs are not known, consequently such genes would be missed by target depending on the identification method [9, 12]. Beyond this, some snoRNAs are shown to target specific mRNAs, suggesting other functions, e.g., interference with A-to-I editing [7, 9, 12–14]. In order to identify C/D box and H/ACA box snoRNAs, snoReport uses position-specific weighted matrices (PWM’s) to identify boxes, together with a set of restrictions related to the secondary structure prediction, usually, restrictions about distance between regions of the secondary structure, and whether it forms the hairpins for H/ACA box snoRNAs, or the loop for a C/D box snoRNA.
SnoReport produced good results. In the test phase, snoReport presented 96 % of sensitivity and 91 % of specificity for the C/D box snoRNA classification, while for H/ACA box snoRNAs, it has shown 78 % of sensitivity and 89 % of specificity. However, snoReport has been trained on almost exclusively mammalian sequences, having used some default parameters for the Support Vector Machine (SVM) classifier. To date, many new sequences of snoRNAs for different vertebrate organisms have been identified, and experimentally confirmed. Furthermore, many tools and databases used to build snoReport have been improved. This suggests that snoReport has to be updated, in order to use new data and refined machine learning techniques to improve its performance.
We improved snoReport, by extracting new features for both box C/D and H/ACA box snoRNAs, developing a more sophisticated technique in the SVM training phase (with recent data from vertebrate organisms and a different approach to refine the C and γ SVM parameters), and using new versions of the tools and databases previously taken to build snoReport. To validate this new version of snoReport, we tested it in different organisms. These experiments have shown a very good performance.
This text is organized as follows. In the next section, we describe the methods used for building the new version of snoReport, particularly, data sources and the new workflow, besides the new features and details of the training phase. After, we show the results obtained by the new version of snoReport with different species of organisms. Following, we discuss these results. Finally, we conclude and suggest future work.
Methods
First, data sources, software components, and the workflow used to build the new snoReport are described. Next, the new attributes for boxes H/ACA and C/D snoRNAs used in the SVM classifier are shown.
Data sources
In snoReport, two datasets were used for the training and testing phases: positive samples and negative samples. The positive sample set was composed of H/ACA box and C/D box snoRNAs, while the negative one was obtained from a dinucleotide shuffling procedure executed in the positive samples with the EDeN [15] library.
The positive sequences from each class of snoRNAs were divided in two datasets, to be used in the learning process. In order to avoid overfitting, these datasets were created such that very similar sequences would not be stored in different datasets. First, we clustered the sequences using ClustalW [16] with criterion nucleotide similarity, which generated 157 clusters for C/D box snoRNA and 101 clusters for H/ACA box snoRNA. After, 10 sequences from distinct vertebrates organisms were extracted from each cluster, noting that clusters containing less than 10 sequences were discarded. Therefore, a consensus sequence from each cluster was obtained with ClustalW and Cons (for EMBOSS [17]), and these sequences were used to generate a distance tree, with the neighbour-joining method [18] from ClustalW2 - phylogeny [19]. The next step was to divide this distance tree in two parts, which allowed to create the two datasets containing similar sequences. The generated trees of C/D box snoRNA and H/ACA box snoRNA clusters can be viewed on Additional file 1.
Table 1 shows the number of sequences of each dataset.
Position-specific weight matrices (PWMs) were used to represent each characteristic sequence motif of H/ACA box and C/D box snoRNAs. These PWMs were obtained by scanning the boxes from snoRNAs of vertebrates. A PWM shows the probability that each nucleotide can be found in a particular position of a box motif. These PWMs generate scores used to identify boxes in a candidate sequence. To create thresholds for each box, we scanned snoRNA sequences with a window size equal to the length of the corresponding box. The scanned candidate boxes that were not true boxes were classified as negative boxes. Thus, we generated a density plot to define the thresholds. Figures 3, 4, 5 and 6 show these density plots.

Density plot of H box PWM-based scores

Density plot of ACA box PWM-based scores

Density plot of C box PWM-based scores

Density plot of D box PWM-based scores
In the validation phase, we used sets of predicted, and partially confirmed with experiments, snoRNAs from many organisms: human [10], nematodes [20], Drosophilids [21], chicken [22], platypus [23] and leishmania [24]. These sequences were manually extracted from Additional files 1, 2 and 3 of each paper (originally in pdf format and doc format tables).
Software components
RNA secondary structure prediction was performed using Vienna RNA Package, current version 2.15, in particular RNAfold [25], RNAz [26] and RNALfold [27]. RNAfold predicts a secondary structure associated with the minimum free energy (MFE) of a single stranded RNA or DNA sequence. RNALfold computes locally stable RNA secondary structure with a maximal base pair span. It was used here in order to find the start position of a H/ACA box snoRNA candidate. RNAz was executed to calculate zscore, an attribute of the feature vector of H/ACA box snoRNA that represents the thermodynamic stability of a ncRNA secondary structure.
Many tools available in the libSVM version 3.20 [28] performed the classification of H/ACA box snoRNA and C/D box snoRNA:
-
grid.py: to identify good values for C and γ SVM parameters;
-
svm-scale: to scale the feature vector;
-
svm-train: to perform training and build a model used for predicting new candidates in the svm-predict tool;
-
svm-predict: to predict sequences not used in the training phase.
In order to calculate different performance measures (not available in libSVM), we developed a script using scikit-learn library [29] to calculate Accuracy, F-score, Average Precision, ROC AUC score and Residual sum of squares (RSS). Using these software components, the snoReport 2.0 was entirely rewritten in the C language.
Identifying snoRNA candidates in genomic sequences
As said before, both classes of snoRNAs, H/ACA box and C/D box, can be distinguished by their characteristic boxes, and some specific secondary structure features. For this, each class of snoRNA has a specific way to searching for candidates, described as follows.
Searching for H/ACA box snoRNAs in a genome sequence was performed with the following steps (Fig. 7):
-
The genome sequence is scanned in order to find potential H boxes with PWM-based scores above a certain threshold;
Fig. 7 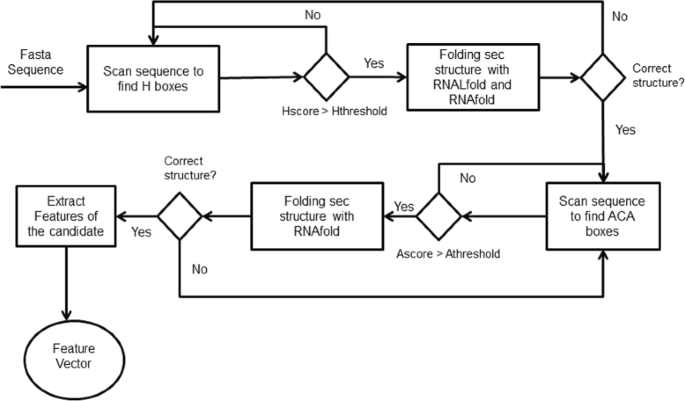
Workflow to identify H/ACA on snoReport 2.0
-
If one H box candidate has a good PWM-based score, we executed first RNALFold to find the start position of one H/ACA box snoRNA candidate, and then RNAfold with some constraints to predict its secondary structure;
-
If the sequence between the start position and the H box candidate has a correct secondary structure, we look for ACA box candidates with a maximum distance of 120 nts and presenting a PWM-based score above a certain threshold;
-
Finally, RNAfold is called for the sequence between H box and ACA box. If this sequence has the correct structure, features for this candidate were extracted.

Restrictions used to predict secondary structure are specific for each class of snoRNA. For the secondary structure of H/ACA box snoRNA, the region upstream of box H and the region between box H and ACA are used to fold into single stem loop structures. In the cell, snoRNA interacts with a set of different proteins that stabilize the large interior loop containing the target binding site. Without these proteins, standard MFE folding algorithms can predict base pairs within this loop. Therefore, to open the target region, we constrained the 14th base upstream of boxes H and ACA, and in most cases the complete interior loop turns out to be unpaired in the MFE structure. Figure 8 shows the canonical representation of H/ACA box snoRNAs.

Canonical secondary structure of H/ACA box snoRNA, with two hairpins and two short-single stranded regions containing boxes H and ACA (located 3 nt upstream of the 3’ end). The hairpin contains bulges, or recognition loops, which form complex pseudoknots with the target RNA, where the target uridine is the first unpaired base [7, 10]
Searching for C/D box snoRNAs in a genome sequence was performed with the following steps (Fig. 9):
-
The genome sequence is scanned in order to find C boxes with PWM-based scores above a certain threshold;
Fig. 9 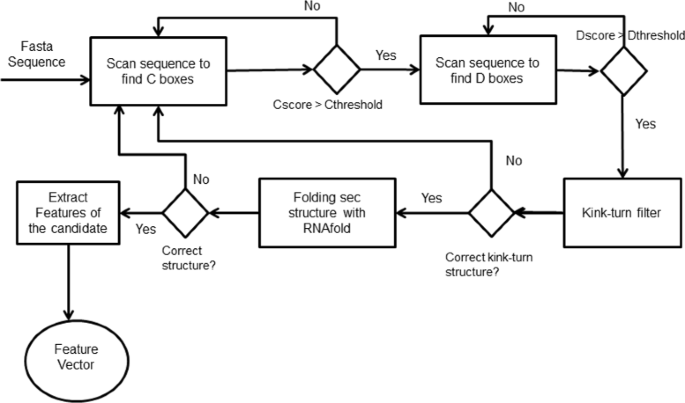
Workflow to identify C/D on snoReport 2.0
-
If the C box candidate has a good PWM-based score, we look for D box candidates with a maximum distance of 200 nts with PWM-based score above a certain threshold;
-
The candidate has its kink-turn structure (kink turn is a structural motif of RNAs that generates a kink in the helical axis [30]) tested, and in case of having the correct one, RNAfold is called to predict its secondary structure;
-
If it has the correct secondary structure, features for this candidate are extracted.

For the secondary structure of C/D box snoRNA, the complete region from the start of box C to the end of box D has to remain unpaired. Many studies have shown that C/D box snoRNAs must have a perfect kink turn structure that boxes C and D [31–33]. For this, snoreport 2.0 has a kink turn structure test, where a C/D box snoRNA candidate must have: G ∙A dinucleotides in box C (RUGAUGA) and box D(CUGA); at least one uridine on the U-U pair (RUGAUGA and CUGA); and a Watson-Crick base pair between the 6th nt of C and the 1st nt of D box (RUGAUGA and CUGA). Figure 10 shows the kink turn structure of C/D box snoRNA, and Fig. 11 shows the canonical representation of C/D box snoRNAs.

Kink turn structure of C/D box snoRNA [31]

Canonical secondary structure of C/D box snoRNA [10]. Boxes C and D are located near to the 5’ and 3’ ends, noting that they are frequently folded together by a short stem. Normally, imperfect copies of C and D boxes, called D’ and C’, are located internally in the loop, ordered as C, D’, C’ and D. The target RNA is guided by antisense elements located upstream of D box or D’ box
Extraction of feature vectors
If a snoRNA candidate meets all the previously described filters, and fold the secondary structure, snoReport 2.0 extracts some attributes from a H/ACA (C/D) box snoRNA candidate, in order to build a feature vector, which will be the input for the Support Vector Machine (SVM). Some changes in the feature vectors of both H/ACA box and C/D box snoRNA candidates were introduced, compared to the previous version of snoReport.
In the feature vector of H/ACA box snoRNA, the following new attributes were included: AC, GU, zscore, Hscore, ACAscore, LloopSC, RloopSC, LloopYC, RloopYC, LloopSym and RloopSym. Table 2 shows all the attributes that have to be extracted from a H/ACA box snoRNA candidate.
The attribute mfeC shows the MFE of folding with constraint nucleotides, providing the information of how much “effort” is needed to force the candidate sequence to fit the requested structure, or if the candidate is more stable in another structure. AC, GC and GU contents are used to distinguish ncRNAs from other RNAs. For example, the human genome has approximately 42 % of GC content, but single sequences of miRNAs and H/ACA box snoRNAs have 50 % of average GC content [34]. The zscore feature is obtained with RNAz [26], representing the thermodynamic stability of a ncRNA secondary structure. Values Hscore and ACAscore were computed using PWMs of H box and ACA box, respectively. Attributes LseqSize, RseqSize, LloopSC, RloopSC, LloopYC, RloopYC, LloopSym and RloopSym help to discriminate arbitrary double stem loop structures from H/ACA stem loop structures.
In the feature vector of C/D box snoRNA, new attributes were also included: zscore, bpStem, l u5,l u3, stemUnpCbox, stemUnpDbox. Table 3 shows the attributes that have to be extracted from a C/D box snoRNA candidate.
Attributes mfeC and mfe are used to distinguish both RNAfold folding procedures, with and without restrictions, respectively. Attributes E avg and E stdv represent average and standard deviation of folding energy for random sequences with identical nucleotide frequency in RNAz. Values Cscore and Dscore were computed using PWMs of C box and D box, respectively. The other attributes (bpStem, l u5, l u3, stemUnpCbox, stemUnpDbox) allow to distinguish C/D box snoRNAs from other RNAs according to the stem found by the secondary structure prediction.
Training and test phases
Figure 12 shows the training and test phases workflow of snoReport 2.0.

Workflow of snoReport 2.0
Since we have two datasets for each class of snoRNA, two different training and test phases were performed, one with dataset 1 as training and dataset 2 as test, and vice versa. For each dataset, negative samples were generated with a dinucleotide shuffling procedure from EDeN. In order to reliably measure the quality of the learning, we repeated the training and test phase 10 times for each dataset, generating on each time new negative samples. After creating the training and test dataset, the feature vector was scaled from -1 to 1, using svm-scale for a better SVM classification.
The next step was to perform a grid search for the C and γ parameters, using grid.py (available in libSVM v3.20), a parameter selection tool for C-SVM classification that uses the RBF (radial basis function) kernel. It uses a cross validation technique (in our case, 10-fold) to estimate the accuracy (another criteria could be used as well) of each combination of C and γ in the specified range, which allowed to choose the best values. Following Hsu [35], “a practical method to identify good parameters is to try exponentially growing sequences of C and γ”. Therefore, we first investigated all the combinations of these two parameters ranging both from 2−15 to 215, shifting 21 for each step of the grid-search (for example, 2−15,2−14,...215). Figure 13 shows an example of a performed grid search.

Grid search using accuracy as a criterion for the C/D box snoRNA classification. Each line represents the accuracy obtained in the training phase, using parameters C and γ with 10-fold cross validation. Here, the green line represents 98.5 % of accuracy using any point of this line
After estimating parameters C and γ, the training phase was performed using svm-train, which used C-SVM with the RBF kernel and probabilities estimates enabled. After training, we obtained a classifier (called model) used as input in svm-predict to predict snoRNAs from sequences not used in the training phase.
For a more refined analysis, we used the scikit-learn library [29], which allowed to obtain three performance measures to better evaluate and compare the snoReport 2.0 with the previous snoReport:‘q
-
Fixed threshold (Accuracy and F-score): a sample is classified as positive if its score (or probability) is above a certain fixed threshold;
-
Dynamic threshold (Average precision – APR – and Area Under the Curve – AUC): measures based on moving thresholds along the positive class. It returns the area under the precision-recall curve (APR) and the area under the ROC curve (AUC);
-
Residual sum of squares (RSS): shows the discrepancy between data and an the estimator model.
Results
First, we present statistics of the performance tests. Then we will discuss the results of executing snoReport 2.0 on real data of different organisms.
Statistics
To identify H/ACA box and C/D box snoRNAs, we built two different datasets for each class of snoRNAs. For the learning phases, we used one dataset as training and the other for test (and vice versa). Each training was repeated 10 times, and our results show the average of the obtained results, together with their corresponding standard deviation. Tables 4 and 5 show the results of the test phase of each snoRNA class obtained with snoReport 2.0.
In order to compare the results with the previous version of snoReport, we executed on snoReport 1.0 the datasets used in the tests with snoReport 2.0. Tables 6 and 7 show the results. These results have shown that snoReport 2.0 presented a better performance to predict vertebrate data, with all the performance measures above 90 %. For H/ACA box snoRNA, the F-score, which consider both precision and recall, snoReport 2.0 was 10.9 % better, having improved the old version. For C/D box snoRNA, we again see an increase of 14,92 % on F-score, and better performances on all the other measures. Thus, snoReport 2.0 showed a significant improvement compared to the previous version.
Validation on real data
To verify the quality of prediction, validation on real data was performed with two experiments. In the first one, we executed snoReport 2.0 with a set of previously predicted vertebrate and invertebrate sequences, some of them partially confirmed in experiments, in humans, nematodes, drosophilids, platypus, chickens and leishmania. Tables 8 and 9 show the summary of these results in vertebrates and invertebrates organisms, respectively.
Yang et al. [10] identified 54 snoRNAs, 21 C/D box and 32 H/ACA box in human, using snoSeeker, a method based on probabilistic models, pairwise whole-genome alignments of eukaryotes, in which the user can include information of the putative target region or not (to find orphan snoRNAs). The previous version of snoReport predicted 11 out of 21 C/D box snoRNAs and 23 out of 32 H/ACA box snoRNAs, while snoReport 2.0 predicted 21 C/D box snoRNAs and 28 H/ACA box snoRNAs.
Schmitz et al. [23] identified 166 individual snoRNAs in a platypus brain cDNA library, generated from small non-protein-coding RNAs. After, using BLAST searches in platypus genomic sequences, they found 51 more sequences of snoRNA. We predicted 42 out of 144 C/D box snoRNAs, and 45 out of 73 H/ACA box snoRNAs.
Shao et al. [22] identified 132 C/D box snoRNAs in chicken using CDseeker and 69 H/ACA box snoRNAs using ACAseeker (both programs are used in snoSeeker [10]). We predicted, with snoReport 2.0, 112 out of 132 C/D box snoRNAs, and 66 out of 69 H/ACA box snoRNAs.
Zemann et al. [20] used a combination of high-throughput cDNA library screening and computational search strategies to find 121 snoRNAs (168 are shown in their supplementary material) in Caernorhabditis elegans. Our snoReport 2.0 predicted 32 out of 108 C/D box snoRNAs, and 46 out of 60 H/ACA box snoRNAs.
Huang et al. [21] performed a large-scale genome wide analysis to identify both classes of snoRNAs in Drosophila melanogaster using experimental and computational RNomics methods, having found 119 snoRNAs. Our snoReport 2.0 predicted 2 out of 63 C/D box snoRNAs, and 39 out of 56 H/ACA box snoRNAs.
Finally, Liang et al. [24] used a genome-wide screening approach to identify 62 C/D box snoRNAs and 37 H/ACA box snoRNAs of closely related pathogens of Leishmania major. We did not identify any C/D box nor H/ACA box snoRNAs. It is interesting to note that H/ACA box snoRNAs from Leishmania major are quite different from the canonical H/ACA box snoRNAs of yeast and vertebrate. For example, they lack a recognizable H box, presenting an AGA box instead of an ACA box [9]. Our snoReport 2.0 was designed to identify canonical snoRNAs from many different organisms, thus to predict H/ACA box snoRNAs from organisms that are different from the canonical model, we should use a different training set, together with a revision of the attributes of the feature vector.
In the second experiment, we investigated false positives in snoReport 2.0. A variety of ncRNA families were taken from RFAM [36] (100 sequences with sizes compatible to snoRNAs), and a set of 100 randomly chosen genomic loci of snoRNA comparable size taken from human genome GRCh38.p7 [37] chromosomes 15, 16, 21 and 24 (25 sequences from each chromosome). To construct the confusion matrices (Tables 10 and 11), we chose one representative sequence from each cluster of snoRNAs, as described in the data source section, in a total of 224 snoRNAs, 132 C/D box and 92 H/ACA box.
For the C/D box snoRNA experiment, we obtained a precision of 100 %, a recall of 74.2 % and a F-score of 85.2 %. This shows that snoReport 2.0 is reliable to predict true C/D box snoRNAs, since no other kind of ncRNA was predicted as C/D box snoRNA.
For the H/ACA box experiment, we obtained a precision of 93.8 %, a recall of 65.2 % and a F-score of 76.9 %. Analogous to the C/D box snoRNA experiment, our method prevent to obtain false positives, confirming that it is reliable to predict snoRNAs. Regarding the four non H/ACA box snoRNAs identified as so, three sequences belonging to chromosome 15 and one to chromosome 16. It is noteworthy that, in chromosome 15, one unknown H/ACA box snoRNA, with probability of 93 %, was located inside the protein TRPM1 [38]. The other two snoRNAs were located in uncharacterized contigs. In chromossome 16, we identified a H/ACA box snoRNA, with probability of 91 %, inside the uncharacterized LOC102723323 ncRNA [39].
Discussion
In this work, we refined the training phase of the SVM method, using different features in the characteristic vector, more data from different vertebrate organisms, and new versions of the tools and data bases used to build the first version of snoReport. We carefully chose good values for the C and γ SVM parameters using grid searches.
All these steps allowed us to improve the performance of snoReport, avoiding false positives and finding more snoRNAs. H/ACA box snoRNA classifier had an improvement of 10.9 % regarding to F-score, with the same data, when compared to the first version of snoReport. Besides, the high score achieved from average precision,ROC AUC score and RSS show us that the predictions have a high degree of reliability. The same could be observed for C/D box snoRNA classifier, which have an improvement of 14.92 % regarding to F-score, and more than 90 % of all performance measures presented, allowing us to have high rate of quality on each prediction.
The validation phase showed, in the first experiment, that snoReport 2.0 predicted 67.43 % of snoRNAs from vertebrates organisms, which shows that snoReport 2.0 can identify snoRNAs with significantly higher precision while maintaining recall. It is noteworthy that many sequences used for validation was not yet experimentally validated, and maybe some of them can be false positives, or are not representatives of the canonical snoRNAs (like the snoRNAs in leishmania). In this case, snoReport 2.0 could discard these candidates. Since snoReport was trained with vertebrate sequence, snoRNAs in invertebrates could not be detected efficiently by snoReport. To deal with some of these organisms, it is necessary to discover new features that describe those non standard snoRNAs and use particular datasets in machine learning tasks. However, we find 69,64 % and 76,67 % of H/ACA box snoRNAs of nematodes and drosophilids described in literature, which suggests that H/ACA box snoRNA predictor from snoReport can be used with high performance. In the second experiment, the validation confirmed that snoReport 2.0 prevents to prediction of false positives.
Therefore, snoReport 2.0 constitutes a substantial improvent over its first version, and is now more efficient and reliable to identify both classes of snoRNAs. It can be used for many different organisms, even invertebrates, with high quality of prediction.
Conclusion
In this article, we presented snoReport 2.0, a reliable and efficient tool to predict the two main classes of snoRNAs in different organisms. This version is a refinement of a previous version of snoReport, obtained with extensive improvements in the SVM method, and the use of new versions of tools (specially those to predict secondary structures) and databases. In contrast to previous methods for snoRNA identification, snoReport 2.0 can identify both guide and orphan snoRNAs without using any information of putative target sites within ribosomal or spliceosomal RNA nor using multiple alignments. Experiments with very different organisms have shown good performance, even in invertebrates organisms (for H/ACA box snoRNA), showing that snoReport 2.0 can be used to obtain reliable prediction of snoRNAs in a variety of organisms. Besides, it prevents to predict false positives.
Future work include to create specific datasets for different kinds of organisms (e.g, for invertebrates), and to study at what extent different approaches to fold the sequences and different machine learning methods (e.g., using EDeN to transform the secondary structure of snoRNAs in a graph representation, that can be decomposed in a sparse vector) allow to find intrinsic features or even to predict new snoRNAs. Clearly, these techniques could affect the performance of snoReport 2.0. Our method could also be used to identify snoRNAs in specific species, e.g., fungi (Paracoccidioides brasiliensis, Schizosaccharomyces pombe and Pichia pastoris), or to find specific features and perform a SVM training to identify snoRNAs in leishmania. Finally, a general method could be developed to allow SVM training with particular organisms, according to an user’s necessity.
Availability and requirements
-
Project Name: SnoReport v2.0;
-
Project home page: http://www.biomol.unb.br/snoreport;
-
Operation system(s) Linux;
-
Programming language C ansi;
-
Other requirements: Vienna RNA Package v2.1.5 (particularly RNAfold, RNALFold and RNAz);
-
License: GNU GPL
-
Any restriction to use by non-academics: No restrictions
Abbreviations
- MFE:
-
Minimum free energy
- ncRNA:
-
Non-coding RNA
- PWMs:
-
Position-specific weight matrices
- snoRNA:
-
Small nucleolar RNA
- SVM:
-
Support Vector Machine
References
Machado-Lima A, et al. Computational methods in noncoding RNA research. J Math Biol. 2008; 56(1–2):15–49.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011; 12:861–74.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–7.
Collins LJ. Characterizing ncRNAs in human pathogenic protists using high-throughput sequencing technology. Front Genet. 2011; 2:96.
Yongsheng L, Yunpeng Z, Shengli L, Jianping L, Juan C, Yuan W, Yixue L, Juan X, Xia L. Genome-wide DNA methylome analysis reveals epigenetically dysregulated non-coding RNAs in human breast cancer. Sci Rep. 2015; 5(8790):1–12.
Lluch-Senar M, Delgado J, Chen WH, Lloréns-Rico V, O’Reilly FJ, Wodke JAH, Unal EB, Yus E, Martinez S, Nichols RJ, Ferrar T, Vivancos A, Schmeisky A, Stülke J, van Noort V, Gavin AC, Bork P, Serrano L. Defining a minimal cell: essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol Syst Biol. 2015; 11(780):1–7.
Falaleeva M, Stamm S. Processing of snoRNAs as a new source of regulatory non-coding RNAs. BioEssays. 2013; 35(1):46–54.
Stepanov GA, Filippova JA, Komissarov AB, Kuligina EV, Richter VA, Semenov DV. Regulatory Role of Small Nucleolar RNAs in Human Diseases. BioMed Res Int. 2015; Article ID 206849:1–10.
Hertel J, Hofacker IL, Stadler PF. SnoReport: computational identification of snoRNAs with unknown targets. Bioinformatics. 2008; 24(2):158–64.
Yang J, et al. snoSeeker: an advanced computational package for screening of guide and orphan snoRNA genes in the human genome. Nucleic Acids Res. 2006; 34(18):5112–23.
Lestrade L, Weber MJ. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006; 34(suppl 1):158–62.
Kim SH, et al. Plant U13 orthologues and orphan snoRNAs identified by RNomics of RNA from Arabidopsis nucleoli. Nucleic Acids Res. 2010; 38(9):3054–67.
Vitali P, Basyuk E, Le Meur E, Bertrand E, Muscatelli F, Cavaillé J, Huttenhofer A. ADAR2-mediated editing of RNA substrates in the nucleolus is inhibited by C/D small nucleolar RNAs. J Cell Biol. 2005; 169(5):745–53.
Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006; 311(5758):230–2.
Costa F, Grave KD. Fast neighborhood subgraph pairwise distance kernel In: Wrobel S, Fürnkranz J, Joachims T, editors. Proceedings of the 26th International Conference on Machine Learning: 2010. p. 255–62.
Thopmson J, Higgins DGG, Gibson T. Clustalw: improving the sensitivity if progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994; 22:4673–80.
Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite,. Trends Genet TIG. 2000; 16(6):276–7.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987; 4(4):406–25.
McWilliam H, et al. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013; 41(Web Server issue):597–600.
Zemann A, et al. Evolution of small nucleolar RNAs in nematodes. Nucleic Acids Res. 2006; 34(9):2676–685.
Huang ZP, Zhou H, He HL, Chen CL, Liang D, Qu LH. Genome-wide analyses of two families of snoRNA genes from Drosophila melanogaster, demonstrating the extensive utilization of introns for coding of snoRNAs. RNA. 2005; 11(8):1303–16.
Shao P, et al. Genome-wide analysis of chicken snoRNAs provides unique implications for the evolution of vertebrate snoRNAs. BMC Genomics. 2009; 10:86.
Schmitz J, et al. Retroposed SNOfall - A mammalian-wide comparison of platypus snoRNAs. Genome Res. 2008; 18:1005–10.
Liang XH, et al. A genome-wide analysis of C/D and H/ACA-like small nucleolar RNAs in Leishmania major indicates conservation among trypanosomatids in repertoire and in their rRNA targets. Eukaryot Cell. 2007; 6:361–77.
Hofacker IL, Fontana W, Stadler PF, Bonhoeffer SL, Tacker M, Schuster P. Fast Folding and Comparison of RNA Secondary Structures. Monatsh Chem. 1994; 125:167–88.
Gruber AR, et al. RNAz 2.0: Improved Noncoding RNA Detection. In: Pacific Symposium on Biocomputing: 2010. p. 69–79.
Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003; 31(13):3429–31.
Chang CC, Lin CJ. LIBSVM: a library for Support Vector Machines. 2001. Software available at: http://www.csie.ntu.edu.tw/%7Ecjlin/libsvm/. Accessed 13 July 2016.
Pedregosa F, et al. Scikit-learn: Machine learning in Python. J Mach Learn Res. 2011; 12:2825–30.
Klein DJ, Schmeing TM, Moore PB, Steitz TA. The kink-turn: a new rna secondary structure motif. EMBO J. 2001; 20(15):4214–21.
Bartschat S, et al. snoStrip: a snoRNA annotation pipeline. Bioinformatics. 2014; 30(1):115–6.
Watkins NJ, et al. A common core RNP structure shared between the small nucleolar box C/D RNPs and the spliceosomal U4 snRNP. Cell. 2000; 103(3):457–66.
Xia L, Watkins NJ, Maxwell ES. Identification of specific nucleotide sequences and structural elements required for intronic U14 snoRNA processing. RNA. 1997; 3(1):17–26.
Washietl S, et al. Structured RNAs in the ENCODE selected regions of the human genome. Genome Res. 2007; 17(6):852–64.
Hsu C, Chang C, Lin C. A practical guide to support vector classification. 2010. https://www.csie.ntu.edu.tw/%7Ecjlin/papers/guide/guide.pdf. Accessed 19 May 2016.
Burge SW, et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013; 41(Database-Issue):226–32.
NCBI GRCh38.p7. 2016. Available at: http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.33. Accessed 13 July 2016.
Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, Tepper RI, Shyjan AW. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 1998; 58(7):1515–20.
NCBI LOC102723323. 2016. Available at: http://www.ncbi.nlm.nih.gov/gene/?term=LOC102723323. Accessed 13 July 2016.
Acknowledgements
J.V.A. Oliveira has been supported by CAPES scholarship. M.E.M.T. Walter has been continuously supported by productivity fellowship from CNPq (project 308509/2012-9).
Declarations
This article has been published as part of BMC Bioinformatics Volume 17 Supplement 18, 2016. Proceedings of X-meeting 2015: 11th International Conference of the AB3C + Brazilian Symposium on Bioinformatics: bioinformatics. The full contents of the supplement are available online https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-17-supplement-18.
Funding
Publication charges for this article was funded by University of Brasilia (UnB).
Availability of data and material
All software implementations and datasets are publicly available on http://www.biomol.unb.br/snoreport.
Authors’ contributions
JVAO proposed the new approach of snoReport, implemented the snoReport 2.0, and performed the SVM test and validation phases. JH identified the attributes of the feature vectors of both H/ACA box snoRNA and C/D box snoRNA, and provided data for the training and test phases. FC and RB supported with the machine learning analyses and the refinement of the dataset. MEMTW and PFS supported the proposal and discussion of the results of the method. All authors wrote the text. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1
Trees of C/D box snoRNAs and H/ACA box snoRNAs. The generated distance trees of C/D box snoRNA and H/ACA box snoRNA clusters, used to build the datasets for the training and testing phases. (PDF 47 kb)
Additional file 2
PWMs to identify snoRNA boxes. Position-specific weight matrices (PWMs) used to identify boxes of both classes of snoRNAs. (PDF 64 kb)
Additional file 3
Machine learning statistics. Statistics of all learning procedures made. (ODS 61 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
de Araujo Oliveira, J., Costa, F., Backofen, R. et al. SnoReport 2.0: new features and a refined Support Vector Machine to improve snoRNA identification. BMC Bioinformatics 17 (Suppl 18), 464 (2016). https://doi.org/10.1186/s12859-016-1345-6
Published:
DOI: https://doi.org/10.1186/s12859-016-1345-6