- Software
- Open access
- Published:
FEDRO: a software tool for the automatic discovery of candidate ORFs in plants with c →u RNA editing
BMC Bioinformatics volume 20, Article number: 124 (2019)
Abstract
Background
RNA editing is an important mechanism for gene expression in plants organelles. It alters the direct transfer of genetic information from DNA to proteins, due to the introduction of differences between RNAs and the corresponding coding DNA sequences. Software tools successful for the search of genes in other organisms not always are able to correctly perform this task in plants organellar genomes. Moreover, the available software tools predicting RNA editing events utilise algorithms that do not account for events which may generate a novel start codon.
Results
We present Fedro, a Java software tool implementing a novel strategy to generate candidate Open Reading Frames (ORFs) resulting from Cytidine to Uridine (c→u) editing substitutions which occur in the mitochondrial genome (mtDNA) of a given input plant. The goal is to predict putative proteins of plants mitochondria that have not been yet annotated. In order to validate the generated ORFs, a screening is performed by checking for sequence similarity or presence in active transcripts of the same or similar organisms. We illustrate the functionalities of our framework on a model organism.
Conclusions
The proposed tool may be used also on other organisms and genomes. Fedro is publicly available at http://math.unipa.it/rombo/FEDRO.
Background
In mitochondria and chloroplasts of flowering plants the direct transfer of genetic information from DNA to proteins is corrupted by mechanisms that produce primary nucleotide sequences different from the original DNA sequences, by altering the structure of RNA transcripts. The most common among these mechanisms is post-transcriptional mRNA editing, consisting in enzymatic modification of nitrogenous bases, almost exclusively c→u transformation [1]. Most RNA editing events are found in the coding regions of mRNAs and usually at the first and second position of codons, so that the corresponding amino acid is often different from that specified by the unedited codon [2]. Editing can also create new start and stop codons [3, 4] and it can occur in introns [5] and in other non-translated regions [6]. The use of editing to generate aug start codons is well described for plants chloroplasts [7, 8], but there is evidence of it also in plants mithocondria [9]. This phenomenon may represent another level of regulatory control of gene expression. Indeed, the introduction of a translational start codon can make an mRNA accessible for protein synthesis [1]. Within this general context, in plant mitochondria RNA editing proves essential for gene expression. In many cases this mechanism completes the genomic information and it is important to the creation of a functional ORF [10].
The mechanism of RNA editing in plant mitochondria makes it harder studying the transfer of genetic information from DNA to proteins, due to the intervening differences occurring between RNAs and their coding DNA sequences. For this reason, common software tools for gene search helpful in finding canonical genes often fail short in discovering genes in plants and therefore some mitochondrial proteins may remain unknown. Accordingly, complete sequencing of mtDNA of many organisms allowed the identification of canonical genes, but much of the informational content of plant mitochondrial genomes remains still undiscovered. Therefore, finding plant mitochondrial proteins and understanding how they integrate into metabolic and signaling pathways, yet represents a major challenge in cell biology.
Here we present FEDRO, a Java software tool implementing a methodology based on the simulation of c→u RNA editing in the mitochondria of plants. The end result is the prediction of proteins which have been not yet discovered and annotated [11]. Indeed, plant mitochondria may use the editing mechanism on crucial sites, causing the generation of new, i.e., edited, start codons aug from acg. FEDRO is a three-steps approach that first generates a collection of potential ORFs, that cannot be detected by classical discovery techniques, based on the RNA editing simulation of edited starting codons. Then the ORFs which are already known to be encoded as proteins in the input organism are filtered out. The final step is a comparison of the remaining sequences against the BLAST database according to the programs described in [12], in order to select only those potential ORFs which present high sequence similarity with proteins in other organisms, or with transcripts in the same or different organisms.
We illustrate the functionalities of FEDRO on the mtDNA of Oryza sativa, where previous studies systematically identified mRNA editing sites of known ORFs (e.g., [13]) but they did not investigate the possible generation of novel ORFs by the editing of the first codon. FEDRO allows to take into account this aspect, leading to the prediction of novel putative ORFs encrypted via c→u editing.
Implementation
The proposed system aims to automatically simulate the editing mechanisms possibly causing the presence of proteins that are not imputable to ORF sequences obtained by traditional methods (e.g., ORF FINDER [14], STARORF [15]). This is rather meaningful in plants, as already discussed in the Introduction, since mtDNA editing mechanisms can involve nucleotide triplets leading to start and stop codons. In particular, we aim to generate putative proteins not yet discovered in a given input organism. The by far most frequent nucleotide substitution caused by editing is c→u at the RNA level, that is, c→t if we refer to mtDNA. Therefore we consider only this type of nucleotide substitution in our analysis.
In particular, the proposed framework FEDRO consists of three main steps, graphically illustrated in Fig. 1:
-
Extraction: ORFs extraction by editing simulation.
Fig. 1 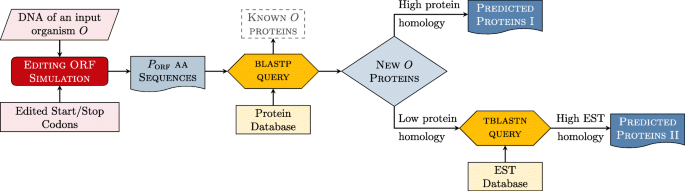
The protein prediction method based on editing simulation
-
Filtering: comparison of the extracted ORFs with proteins already known in the same organism.
-
Selection: selection of the non-filtered ORFs that show high homology with known proteins in other orgnanisms, and search of known transcripts in the remaining ORFs.

Figure 2 shows a screenshot of the main window of FEDRO. In the following we describe in detail each of the involved steps.

A screenshot of the main window of FEDRO
Extraction
This phase is devoted to extract candidate ORFs from the input mtDNA sequence. We aim to extract both ORFs identified by standard start/stop codons and ORFs generated by the c→t editing mechanism using simulation. It is worth pointing out that editing events on the start/stop codons may affect the entire process of ORFs extraction. That is, although FEDRO returns also ORFs identified by standard start/stop codons, they could be different from those returned by ORF FINDER [14] or STARORF [15], due to different cuts of the input mtDNA sequence.
INPUT:
-
the mtDNA sequence of an organism to be analyzed;
-
user-defined threshold θlen: the minimum length of extracted ORFs, equal to 300 by default;
-
considering edited start codons (yes/no), set to yes by default;
-
considering edited stop codons (yes/no), set to no by default;
METHOD: The simplest way to find genes in a genome is that of scanning the input nucleotide sequence by considering all its three possible reading frames, then searching for nucleotide triplets corresponding to start codons and stop codons on a frame, and cutting the sub-sequences comprised between a start and a stop codon, that are the Open Reading Frame sequences. An ORF sequence is considered to be a potential protein encoding segments if its length is at least 300 nucleotides. In particular, there exist one start codon, that is, atg and three stop codons, that are tag,tga and taa. Although ORF sequences can be easily searched for in a genomic sequence by using existing software tools, such as for example ORF FINDER [14] and STARORF [15], these tools do not take into account the role of possible editing processes which could have modified the involved sequences.
Our system simultaneously looks for atg start codons and acg edited start codons. Next, it looks for a stop codon and extracts the ORF if and only if its length amounts at least at the minimum user-defined threshold θlen. Note that, since the stop codon does not have other possible interpretations, it cannot be ignored in order to find largest ORFs. All that given, two issues deserve attention:
-
1
that possible further start codons between the current start codon and the stop codon are ignored;
-
2
that edited stop codons are not taken into account.
The first issue is related to the fact that a start codon can be also be interpreted as amino-acid and using a preceding start codon just generates larger ORFs. Let \(\overrightarrow {\delta }\) be the position where a start codon begins and let \(\overleftarrow {\delta }\) be the position where a stop codon begins. Assume that the number of basis between \(\overrightarrow {\delta }\) and \(\overleftarrow {\delta }\) is larger than θlen. Consider now a possible further start codon beginning in a position δ between \(\overrightarrow {\delta }\) and \(\overleftarrow {\delta }\). Taking δ as the start codon beginning position leads to consider ORF comprised between δ and \(\overleftarrow {\delta }\), which is strictly included in the ORF comprised between \(\overrightarrow {\delta }\) and \(\overleftarrow {\delta }\). Thus, ignoring intermediate start codons, like the one beginning at δ, implies to extract larger ORFs. As shown in the experiments, this does not lead the system to be less accurate.
The second issue is related to the fact that a stop codon does not have other possible interpretations and then each stop codon has to be considered to extract an ORF. Considering editing codons enlarges the set of stop codons to be taken into accounts and thus, leads to the generation of smaller ORFs.
OUTPUT: The output of this phase is a set of candidate ORFs, translated into amino-acid sequences.
Filtering
Since the end goal of our system is to support the discovery of possible novel proteins, the goal of this phase is to filter out from the candidate ORF set, that set of ORFs which is already known to be encoded as proteins in the input organism.
INPUT: The set of candidate ORFs as returned by the extraction phase.
METHOD: To perform this operation, the system queries the protein database by exploiting the web services offered by the BLAST framework and, in particular, the system uses the blastp software to measure the similarity between a candidate and the known proteins, such that two proteins are considered similar if the resulting bitscore is larger than or equal to 200. From the analysis of the returned similarity, three cases can arise:
-
1
the ORF is similar to a protein already known in the input organism;
-
2
the ORF is similar to a protein already known in another organism;
-
3
the ORF is not similar to any already known protein.
Thus, this phase partitions the input set of ORFs in three sets:
-
ORFs encoding proteins already known in the input organism (to-drop set);
-
ORFs encoding proteins already known in other organisms;
-
ORFs non-encoding known proteins.
OUTPUT: This phase outputs the two sets b) and c) of non-dropped ORFs.
Selection
The goal of this phase is to select the set of ORFs to be returned for further analysis by domain experts.
INPUT: Two sets of candidate ORFs.
METHOD: The system returns the set of ORFs encoding proteins already known in other organisms. These ORFs are potentially useful for the prediction of putative proteins in the organism under analysis.
Next, the system takes into account the set of ORFs non-encoding known proteins. New proteins could belong to this set. In order to select promising candidates, the system queries the EST database by exploiting, again, the web services offered by the BLAST framework and, in particular, the system uses the tblastn software. The goal is to look for the presence of transcripts that may be indicative for a transcription of the ORFs sequence in a novel protein.
OUTPUT: Two sets of ORFs to be analyzed by domain experts.
Results
To provide example of application, we show the results returned by FEDRO on the mtRNA of the model plant Oryza sativa (version BA000029.3).
As a first test, we extract all the possible ORF sequences from the considered mtDNA by considering only non-edited start and stop codons (with the constraint of a length equal to or larger than 300), as it is returned by a standard tool for ORFs extraction. Then, we do the same, but by applying FEDRO on the mtDNA of the input organism.
The result is that FEDRO generates standard ORFs which are not affected by editing on the start/stop codons, sometimes as subsequences or intersections with standardly observed ORFs. Most importantly, 43 out of 213, that is, 20% of the candidate ORFs returned by FEDRO cannot be extracted by standard tools, such as ORF FINDER [14] or STARORF [15], which do not consider editing events on the start/stop codons.
The second type of analysis aims at understanding if these putative ORFs result to be significant after the validation step, that is, they present high sequence similarity with known proteins or transcripts. In such a case, they can be put in a final catalogue of predictions to be provided to biologists for further lab inspections. In particular, Tables 1 and 2 show the results of the blastp queries for protein sequence similarity search. Results are sorted with respect to the BLAST bitscore values (also other BLAST scores can be taken into account analogously, such as e-value, percentage of coverage and percentage of identity; we omit them since the corresponding results do not present significant differences with respect to those presented here). The column ORF TYPE highlights if the corresponding ORF is contained in, or intercepts, another ORF returned by a standard tool (sub, int, respectively), or if it is not returned by a standard tool (new). Tables 3 and 4 are analogous to the previous ones, but they contain the putative ORFs resulting from tblastn query for EST sequence similarity search.
The data displayed in the abovementioned tables show that FEDRO is able to successfully generate novel putative ORFs. They constitute a useful catalogue for the biologist in order to identify interesting sequences which may be associated to coding regions, but which cannot be recognized by standard ORFs extraction due to the presence of editing that modifies the ORF sequence start/stop codons.
Conclusions
We propose FEDRO, a Java software tool implementing a novel strategy to generate candidate ORFs resulting from c→u editing substitutions which occur in the mitochondrial genome (mtDNA) of a given input plant. This is useful in order to predict putative proteins of plants mitochondria that have not been yet annotated.
We applied FEDRO on the mtDNA of Oryza sativa, suggesting a set of 45 novel putative ORFs to be verified by experts.
FEDRO may be usefully applied to single out informative subsequences also in other, less studied, organisms. With the catalogue of novel putative ORFs in hand, the biologist can perform further analysis and experiments in laboratory to discover the presence of novel proteins in plants, where the editing mechanisms alter the structure of RNA transcripts.
As our future work, we plan to apply our method on the chloroplast genome of plants, where the c→u editing also occurs. Moreover, we are working on the design of a methodology aiming at identifying possible coding exons of trans-spliced genes in the predicted ORFs. We will also investigate the chance to add alternative initial codons ORF [16] as a significant extra feature of the proposed system. Finally, another interesting direction of investigation regards a context-based analysis of the editing sites, which are already known and annotated for many model organisms. To this aim, both sequence motifs or k-mers based analysis may be considered (see, for example, [17, 18] and [19, 20], respectively) and approaches able to characterize anomalous contexts [21, 22].
Availability and requirements
FEDRO is publicly available at http://math.unipa.it/rombo/FEDRO. As specified in the ’readme.txt’ file, the two files ’FEDRO.jar’ and ’data.properties’ have to be included in the same folder. Moreover, the blastp and tblastn executable files have to be provided, and the ’executable’ field of the ’data.properties’ file has to be updated with their paths.
Abbreviations
- BLAST:
-
Basic local alignment search tool
- EST:
-
Expressed sequence tags
- mtDNA:
-
mitochondrial DNA
- ORF:
-
Open reading frames
References
Takenaka MDDV, van der Merwe JA, Zehrmann A, Brennicke A. The process of RNA editing in plant mitochondria. Mitochondrion. 2008; 8:35–46.
Gray MW, Hanic-Joyce PJ, Covello PS. Transcription, processing and editing in plant mitochondria. Annu Rev Plant Physiol Plant Mol Biol. 1992; 43:145–75.
Hoch B, Maier RM, Appel K, Igloi GL, Kossel H. Editing of a chloroplast mRNA by creation of an initiation codon. Nature. 1991; 353:178–80.
Wintz H, Hanson MR. A termination codon is created by RNA editing in the petunia mitochondrial atp9 gene transcript. Curr Genet. 1991; 19:61–4.
Brennicke A, Marchfelder A, Binder S. RNA editing. FEMS Microbiol Rev. 1999; 23:297–316.
Schuster W, Unseld M, Wissinger B, Brennicke A. Ribosomal protein S14 transcripts are edited in Oenothera mitochondria. Nucleic Acids Res. 1990; 18:229–33.
Kossel H, Hoch B, Igloi GL, Maier RM, Ruf S. CEditing C rpl2 Transcript from Maize Chloroplasts In: Nierhaus KH, Franceschi F, Subramanian AR, Erdmann VA, Wittmann-Liebold B, editors. The Translational Apparatus. Boston: Springer: 1993.
Neckermann K, Zeltz P, Igloi GL, Kossel H, Maier RM. The role of RNA editing in conservation of start codons in chloroplast genomes. Gene. 2007; 146(2):177–82.
Kadowaki K, Ozawa K, Kazama S, Kubo N, Akihama T. Creation of an initiation codon by RNA editing in the coxI transcript from tomato mitochondria. Curr Genet. 1995; 28(5):415–22.
Regina TMR, Lopez L, Picardi E, Quagliariello C. Striking differences in RNA editing requirements to express the rps4 gene in magnolia and sunflower mitochondria. Gene. 2002; 286:33–41.
Fassetti F, Giallombardo C, Leone O, Palopoli L, Rombo SE, Saiardi A. Discovering new proteins in plant mitochondria by RNA editing simulation. In: Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS. SciTePress: 2016. p. 182–189. ISBN 978-989-758-170-0.
Altschul SF, et al.Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25(17):3389–402.
Notsu Y, Masood S, Nishikawa T, Kubo N, Akiduki G, Nakazono M, Hirai A, Kadowaki K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol Genet Genomics. 2002; 268(4):434–45.
Stothard P. The sequence manipulation suite: Javascript programs for analyzing and formatting protein and dna sequences. Biotechniques. 2000; 28:1102–4.
MIT. 2007. http://web.mit.edu/star/orf/. StarORF, Open Reading Frame Finder Tool.
Raczynska KD, Le Ret M, Rurek M, Bonnard G, Augustyniak H, Gualberto JM. Plant mitochondrial genes can be expressed from mRNAs lacking stop codons. FEBS Lett. 2006; 580(24):5641–6.
Furfaro A, Groccia MC, Rombo SE. 2D Motif Basis Applied to the Classification of Digital Images. Comput J. 2017; 60(7):1096–1109.
Pizzi C, Ornamenti M, Spangaro S, Rombo SE, Parida L. Efficient Algorithms for Sequence Analysis with Entropic Profiles. IEEE/ACM Trans Comput Biol Bioinforma. 2018; 15(1):117–28.
Giancarlo R, Rombo SE, Utro F. Epigenomic k-mer dictionaries: shedding light on how sequence composition influences in vivo nucleosome positioning. Bioinformatics. 2015; 31(18):2939–46.
Giancarlo R, Rombo SE, Utro F. In vitro vs in vivo compositional landscapes of histone sequence preferences in eucaryotic genomes. Bioinformatics. 2018; 34(20):3454–60.
Angiulli F, Fassetti F, Manco G, Palopoli L. Outlying property detection with numerical attributes. Data Min Knowl Disc. 2017; 31(1):134–63.
Angiulli F, Fassetti F, Palopoli L. Discovering Characterizations of the Behavior of Anomalous Subpopulations. IEEE Trans Knowl Data Eng. 2013; 25(6):1280–92.
Acknowledgements
All the Authors are grateful to the anonymous Referees, whose comments and suggestions allowed to improve the quality of this manuscript.
Funding
This article did not receive sponsorship for publication. Research by F. F., L. P. and S.E.R. has been partially supported by the projects INDAM GNCS 2017 “Algoritmi e tecniche efficienti per l’organizzazione, la gestione e l’analisi di Big Data in ambito biologico” and GNCS 2018 “Elaborazione ed analisi di Big Data modellati come grafi in vari contesti applicativi”.
Availability of data and materials
The proposed software tool and some example input datasets are available at http://math.unipa.it/rombo/FEDRO.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 20 Supplement 4, 2019: Methods, tools and platforms for Personalized Medicine in the Big Data Era (NETTAB 2017). The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-20-supplement-4.
Author information
Authors and Affiliations
Contributions
FF and SER coordinated the organization and writing of this manuscript. AS proposed the main idea behind the presented approach. OL carried a manually based validation of previous versions of the framework, which was essential to achieve the last proposed one. FF, CG, LP and SER worked to the design and development of the framework, and to its experimental validation. CG is responsible for the implementation of Fedro. All the authors proofread and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fassetti, F., Giallombardo, C., Leone, O. et al. FEDRO: a software tool for the automatic discovery of candidate ORFs in plants with c →u RNA editing. BMC Bioinformatics 20 (Suppl 4), 124 (2019). https://doi.org/10.1186/s12859-019-2696-6
Published:
DOI: https://doi.org/10.1186/s12859-019-2696-6